Perioral pigmentation: What is your diagnosis?
Published Web Location
https://doi.org/10.5070/D30h97t2z6Main Content
Perioral pigmentation: What is your diagnosis?
Santos Patrícia1, Neto Cláudia2, Machado Susana3, Lobo Inês3, Soares José4, Selores Manuela3
Dermatology Online Journal 14 (11): 16
1. Pediatric resident of Hospital Pedro Hispano2. Pediatric resident of Hospital Geral de Santo António (HGSA)
3. Dermatology service of HGSA
4. Gastroenterology service of HGSA, Portugal. patriciajuliopt@yahoo.com.br
Abstract
Pigmented spots in the skin and mucosa (lentigines) can be found in various diseases called familial lentiginosis syndromes; Peutz-Jeghers syndrome (PJS) is one of them. It is characterized by the association of mucocutaneous melanin pigmentation and hamartomatous gastrointestinal polyps. Patients with PJS are at increased risk of intussusception and cancer development (gastrointestinal and non-gastrointestinal tumors). We present a 5-year-old girl with pigmented macules of perioral and perinasal skin, lips, and buccal mucosa and review lentiginoses and the surveillance of PJS.
Introduction
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disease with variable penetrance and clinical heterogenecity [1]. Its incidence is 1 per 120,000 newborns without a racial or sexual predilection [2]. It is characterized by the association of mucocutaneous melanin pigmentation, more often located in the perioral region, and hamartomatous polyps, most commonly in the gastrointestinal tract. Hamartomatous polyps may also occur within the lung, urinary tract, and nasal passages [3].
Germline mutations of LKB1 or the serine threonine kinase 11 (STK11/LKB1) gene located at 19p13.3 have been identified as responsible for PJS [2].
Case report
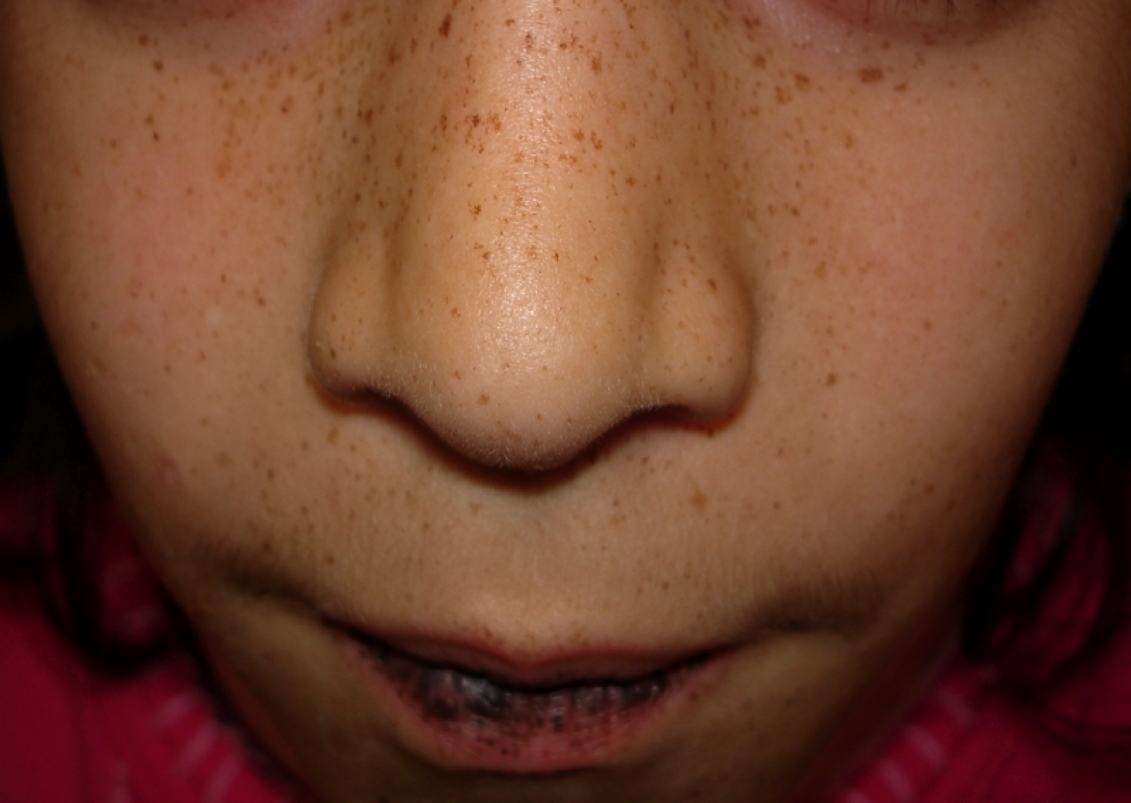
A 5-year-old girl presented for pediatric dermatology consultation with a history of pigmented, round macules of 1-3 mm on her perioral and perinasal skin, lips and buccal mucosa (Figs. 1 & 2) with progressive evolution since the age of two.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. The child face shows pigmented, round macules of 1-3 mm on perioral, perinasal skin and lips. Figure 2. The child buccal mucosa shows pigmented, round macules of 1-3mm. | |
She had no history of abdominal pain, intussusception, intestinal bleeding, rectum prolapse, obstipation, weight loss or asthenia.
On physical examination she was a well-developed girl. There were no pigmented spots on other locations. Abdominal examination did not reveal tenderness, masses or organomegaly. No other abnormalities were noticed on physical examination. She had psychomotor development adequate for her age.
Familial history
The family history revealed that the father had perioral pigmented spots since childhood without gastrointestinal complaints and the younger sister had pigmented macules on the nose and lower lip since 10 months of age.
Laboratory findings
 |  |
| Figure 3a | Figure 3b |
|---|---|
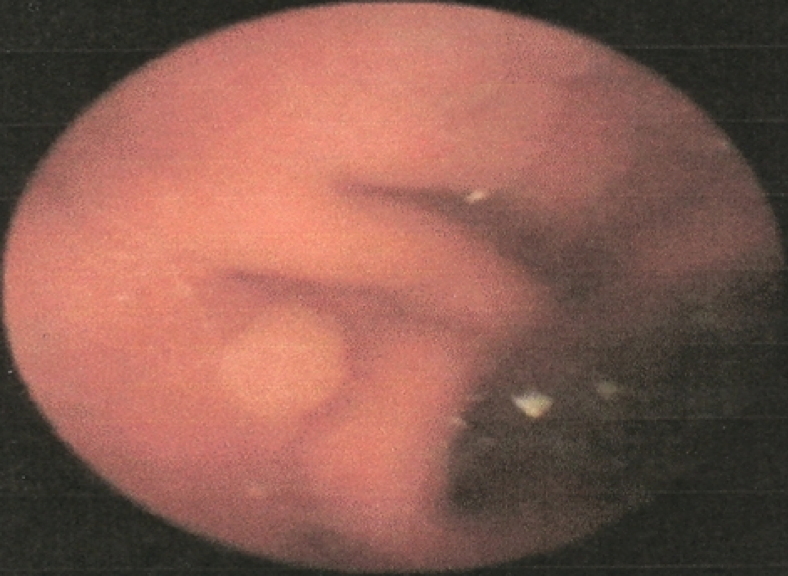
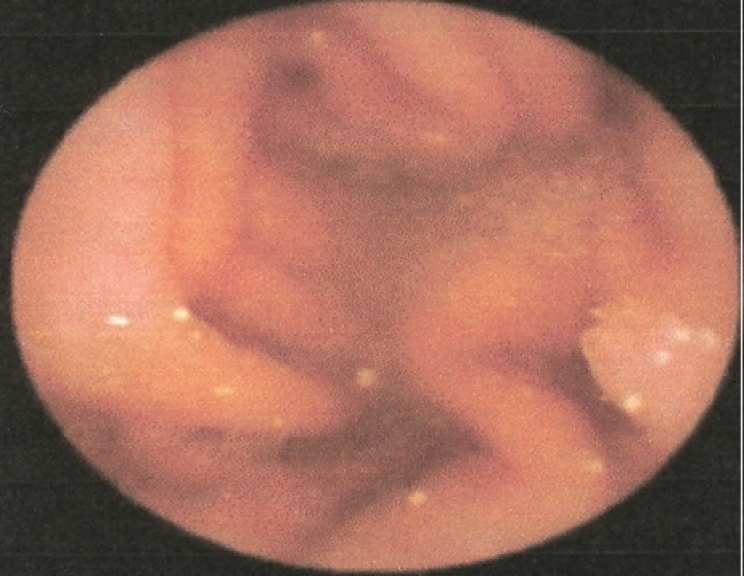
| Figure 3a & 3b. Endoscopic appearance of polyps on the stomach and duodenum | |
Complete blood count was normal. Results of thyroid, renal, and liver function were normal. Echocardiogram and evocative auditory potentials were normal. Genetic study revealed mutation of the STK11 gene at exon 6 and polymorphisms of introns [2, 3, 7]. An upper gastrointestinal, small bowel endoscopies with video-capsule performed at eleven years of age revealed polyps in the stomach, duodenum, jejunum and ileum: these polyps were less than 20 in number and smaller than 5mm (Fig. 3).
Discussion
In PJS pediatric patients the clinical hallmarks are the complications associated with gastrointestinal polyps: abdominal pain, rectal prolapse, intestinal bleeding, anemia, and gastrointestinal intussuception with bowel obstrution [4]. Pigmented macules or lentigines on lips and buccal mucosa can be found in more than 90 percent of individuals with PJS [2] and may be the only clinical finding.
Lentigines may also be a marker of other familial diseases (shown in Table 1). These should be considered in the differential diagnosis [2, 5].
In individuals with PJS, the pigmented macules may fade after puberty and the next clinical finding could be a tumor. Therefore, children presenting with various types of pigmented macules or lentiginosis should be evaluated for possible associated conditions, even if a suspicious family history has not been elicited.
The risk of development of gastrointestinal and extra-intestinal (ovary, endometrium, cervix, breast, testis, lung, pancreas, and proximal bile duct) cancer is increased in PJS. Overall, 50 to 90 percent develop cancer [3] and this development during pediatric years is not negligible [4].
Genetic study of PJS is now possible. The genetic abnormality can be detected in 70 percent of familial cases and 50 percent of sporadic cases [4, 6]. Once diagnosed the child and family must be offered strategies for the prevention of cancer development.
Surveillance should be continued. Actual surveillance guidelines to address the risk of cancer recommend an upper gastrointestinal endoscopy and colonoscopy at 2-year intervals after 10 years of age; clinical appointment/physical examination, hemoglobin, pelvic ultrasound, testes ultrasound, cervical smear every year after puberty; mammography every 2-3 years after 25 years of age and annually after the age of 50 [3, 4, 6].
Treatment depends upon the clinical presentation: laser and intense pulsed light treatments may be successful in treating lentigines on the lips and face; prophylactic endoscopic or laparotomy resection of larger polyps (>15mm) [4]; emergent laparotomy for small-bowel intussusception or obstruction; laparotomy for persistent intestinal bleeding; and adequate treatment of gastrointestinal or extra-intestinal cancers.
Conclusion
Although rare, PJS is a disease with a significant risk of cancer development. The mucocutaneous hyperpigmentation involving the lips and oral cavity should alert physicians to this syndrome. When this sign is present, even without family history or gastrointestinal manifestations, careful evaluation and genetic studies and counseling should be offered to patients and first-degree relatives. Individuals with PSJ should undergo surveillance measures for prevention or early detection of malignancy.
References
1. McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci 2006; 63: 2135-2144. PubMed2. Bishop P, Loftis S, Nowicki M. What Syndrome is This?. Pediatric Dermatology 2004; 21: 503-505. PubMed
3. Girvin F, Glancy S, Dunlop M. Peutz-Jeghers syndrome: A case report and discussion of surveillance recomendations. European Journal of Radiology Extra 62 2007; 81-84.
4. Lopes A, Gonçalves J, Palha A, et al. Peutz-Jeghers Syndrome: Variability of gastrointestinal expression at pediatric age. Acta Med Port 2004; 17: 445-450. PubMed
5. Stratakis CA. Genetic of Peutz-Jeghers Syndrome, Carney Complex and Other Familial Lentiginoses. Horm Res 2000; 54:334-343. PubMed
6. Daniel R, Mcgrath, Spigelman A. Preventive measures in Peutz-Jeghers Syndrome. Familial Cancer. 2001; 1: 121-125. PubMed
© 2008 Dermatology Online Journal