Ulcus cruris associated with prolidase deficiency
Published Web Location
https://doi.org/10.5070/D39v4582j4Main Content
Ulcus cruris associated with prolidase deficiency
Mukaddes Kavala, Ilkin Zindanci, Sibel Sudogan, Zafer Turkoglu, Sukran Sarigul
Dermatology Online Journal 12 (7): 24
Department of Dermatology, Goztepe Training Hospital, istanbul, Turkey. drilkinzindanci@yahoo.com
Abstract
Prolidase deficiency is an autosomal recessive disorder that is associated with chronic cutaneous ulcers, mental retardation, unusual facial appearance, skeletal deformities, joint dislocations, hematological anomalies, splenomegaly, and chronic infections. The most typical finding is chronic, recurrent leg ulcers appearing in early childhood. Prolidase (peptidase-D) is necessary for collagen biosynthesis and its deficiency leads to impairment in connective tissue of the skin, capillaries, and lymphatic vessels. We report a 33-year-old woman who had a 15-year history of nonhealing ulcer on left pretibial region accompanied by splenomegaly, hypochromic microcytic anemia, and thrombocytopenia. Prolidase deficiency is a rare genodermatosis and must be considered in the differential diagnosis of leg ulcers that develop at an early age.
Clinical synopsis
|
|
| Figure 1 | Figure 2 |
|---|---|
|
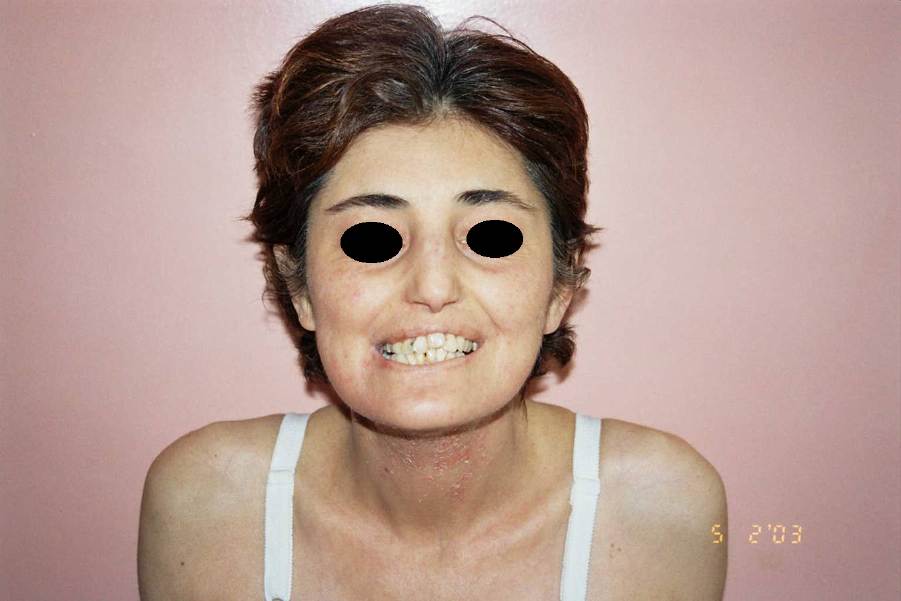
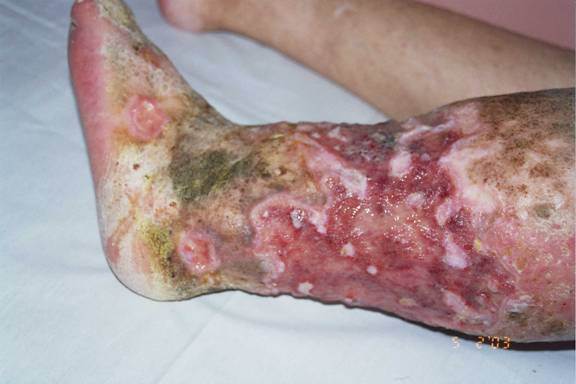
Figure 1. Characteristic facial appearance of the patient Figure 2. Chronic ulcers encircled by atrophic and depigmented cicatricial skin |
|
A 33-year-old woman presented with the complaint of chronic recurrent ulcers on both legs and feet over the course of 18 years. There was no family history of any similar disease. The patient had chronic sinusitis but otherwise was healthy. Examination revealed a characteristic facial appearance with epicanthic folds, a low hair line, frontal bossing, extensive caries, hypoplasia of the jaw, and joint hyperextensibility (Fig. 1). There was edema, induration and extensive irregular ulcers that entirely surrounded the lower third of her left leg (Fig. 2). Laboratory studies, including liver and renal function tests, PPD skin test, rheumatoid factor, C-ANCA, P-ANCA, cryoglobulin screen, antinuclear antibodies, syphilis, and hepatitis B and C serologies were all negative except iron-deficiency anemia and elevated gamma globulin and IgG levels. Prolidase activity in erythrocytes was found to be very low at 19 U/L (normal range 300-900 U/L). Chest radiography, paranasal computed tomogrophic scan, and ophthalmologic examination revealed no abnormality. A skin biopsy showed pronounced fibrosis, neovascularization and perivascular infiltration of neutrophils at the site of acute ulceration. Direct immunofluorescence was negative. The presence of severe ulcerations brought up the differential diagnosis of pyoderma-like lesions associated with Wegener granulomatosis (WG). There was no evidence of systemic involvement or other clinical features to support a diagnosis of WG (the nonspecific histopathologic findings and negative ANCA testing excluded the possibility of WG). The diagnosis of prolidase deficiency was confirmed by reduced prolidase activity in erythrocytes. Treatment with oral manganese sulphate and ascorbic acid, and transfusions of concentrated erythrocytes, and applications of wet dressing resulted in temporary improvement.
Discussion
Prolidase deficiency is a rare, inherited metabolic disorder that results in aminodipeptiduria [1]. Prolidase is an ubiquitous enzyme that plays an important part in collagen degradation and synthesis [2]. Collagen is degraded to aminodipeptides by the action of prolidase. The released proline residues are then recycled for new protein synthesis, and hydroxyproline is excreted in urine. In prolidase deficiency, the enzyme activity is markedly reduced and aminodipeptides cannot be split and excreted into the urine [3, 4]. Prolidase deficiency may be determined by measuring enzyme activity in erythrocytes, leukocytes, or fibroblasts [3]. Clinical manifestations include mental retardation, unusual facial appearance, repeated infections (such as otitis media and sinusitis), skeletal deformities, eye and tooth abnormalities, and extensive skin lesions [4, 5, 6]. The most striking finding is chronic, recalcitrant leg ulcers, which are found in almost all cases [6, 7]. Hyperextensible joints, hypergammaglobulinemia, iron deficiency anemia, and elevated immunoglobulins are other accompanying findings as observed in our case [4, 5]. Ulcers occurring in prolidase deficiency are attributed to the defective recycling of proline residues for the synthesis of new collagen [3, 7]. A variety of treatment methods has been tried with limited and temporary effects [4, 5, 7].
Although our patient had typical features of prolidase deficiency, the differential diagnosis included WG, which has cutaneous involvement as the presenting sign in 9-14 percent of cases. Wegener granulomatosis is a multisystem disease with a predilection for the lung, kidney, and eye [8]. It can present with a limited cutaneous form in which there is no evidence of systemic involvement and negative ANCA testing [9]. The presence of histopathologic findings showing palisading granulomatous inflammation with or without granulomatous vasculitis, and formation of neutrophilic microabscesses with a diffuse c-ANCA values is considered diagnostic of WG [10]. In our case, ANCA testing was repeatedly negative, there was no subsequent development of systemic disease, and no systemic involvement was noted during a 23-month followup; the possibility of WG was excluded by negative ANCA testing together with clinicopathologic findings suggestive of prolidase deficiency.
We suggest that patients who develop chronic and recurrent leg ulcers, even those that resemble other inflammatory disorders, should be examined for prolidase deficiency.
References
1. McKusick VA. In: Mendelian Inheritance in Man. 7th edn. Baltimore: The John Hopkins University Pres. 1986.2. Goodman SI, Solomons CC, Muschenheim F et al. A syndrome resembling lathyrism associated with iminopeptiduria. Am J Med 1968; 45: 152-9.
3. Jackson SH, Dennis AW, Greenberg M: Iminodipeptiduria: A genetic defect in recycling collagen: A method for determining prolidase in erythrocytes. Can Med Assoc J 1975; 113:759-763.
4. Milligan A, Graham-Brown RAC, Burns DA, Anderson I. Prolidase deficiency : a case report and literature review. Br J Dermatol 1989;121:405-9.
5. Ogata A, Tanaka S, Tomoda T et al. Autosomal recessive prolidase deficiency. Three patients with recalcitrant ulcers.Arch Dermatol 1981;117:689-97.
6. Bissonnette R, Friedmann D, Giroux JM et al. Prolidase deficiency: a multisystemic hereditary disorder. J Am Acad Dermatol 1993; 29: 818- 21.
7. Leoni A, Cetta G, Tenni R et al.Prolidase deficiency in two siblings with chronic leg ulcerations. Clinical, biochemical, and morphologic aspects. Arch Dermatol 1987;123:493-9
8. Handfield-Jones SE, Parker SC, Fenton DA, et al. WG presenting as pyoderma gangrenosum. Clin Exp Dermatol 1992; 17: 197-200.
9. Gibson LE, Specks U, Homburger H. Clinical utility of ANCA tests for the dermatologist. Int J Dermatol 2003; 42: 859- 869.
10. Harris NL, Mc Neely WF, Sheppard JO, et al. Case records: Masssachusetts General Hospital N Engl J Med 2002; 346: 1892-1898.
© 2006 Dermatology Online Journal