Dorfman-Chanarin syndrome in two female siblings: A case report and discussion on approach & management
Published Web Location
https://doi.org/10.5070/D39cv1v629Main Content
Dorfman-Chanarin syndrome in two female siblings: A case report and discussion on approach & management
Ram Chander1, Bincy Varghese1, Taru Garg1, Saurabh Mittal1, Smita Singh2
Dermatology Online Journal 17 (4): 7
1. Department of Dermatology & STD, Lady Hardinge Medical College, Shahid Bhagat Singh Marg, New Delhi, India2. Department of Pathology, Lady Hardinge Medical College, Shahid Bhagat Singh Marg, New Delhi, India
Abstract
Dorfman-Chanarin syndrome (DCS) is a very rare disorder of lipid metabolism that exhibits an autosomal recessive pattern of inheritance. Besides ichthyosis, systemic manifestations may be present. We report two female siblings with DCS who presented with non-bullous ichthyosiform erythroderma (NBIE). A peripheral blood smear demonstrated Jordan anomaly. This case emphasizes the need for peripheral blood smear screening in patients with congenital ichthyosis. CASE REPORT: A 2½-year-old female child and her 1-month-old sibling presented with generalized erythema and scaling, which was suggestive of NBIE. Hepatomegaly and ectropion were seen in the older sibling. A peripheral blood smear of both the patients revealed Jordan anomaly. Serum biochemistry revealed abnormal liver function tests, abnormal lipid profile, and elevated muscle-derived enzymes. A diagnosis of Dorfman-Chanarin syndrome was made in both the siblings. Screening for Jordan anomaly in the family members including the parents and maternal and paternal grandmothers was negative. CONCLUSION: The peculiarities in our case include the presence of this disorder in both female siblings along with alopecia in the younger sibling. Hyperlipidemia, noted in one of our cases, is also not a common association. Diagnosing DCS is fairly simple and a high index of suspicion may lead to higher rates of detection of this rare disorder.
Introduction
Dorfman-Chanarin syndrome is a rare metabolic disorder, which is autosomal recessively inherited. It is known by various names including neutral lipid storage disease with ichthyosis (NLSDI) and ichthyosiform erythroderma with leukocyte vacuolation; it is associated with congenital ichthyosis and accumulation of triacylglycerol droplets in most tissues including the liver, muscles, intestinal mucosa, leukocytes and skin fibroblasts [1]. The disorder was first described by Rosenszajn et al in 1966 in two siblings who presented with ichthyosiform erythroderma as the only clinical abnormality [2]. Currently, approximately 40 cases have been reported worldwide, including 5 cases from India [3, 4, 5, 6, 7].
It was only in 2001 that Lefèvre et al first reported CGI-58 mutations to be the cause of DCS [8]. Also known as ABHD-5 (α/β-hydrolase domain-containing protein-5), this gene is present on chromosome 3p21. It is predominantly expressed in the upper granular layers of the epidermis as well as associated with intracellular lipid droplets in tissues such as the neurons and hepatocytes. Activation of Adipose Triglyceride Lipase (ATGL) by CGI-58 brings about catabolism of long chain fatty acids [9]. Thus, a deficiency of CGI-58 leads to an producing to the various clinical manifestations.
Case report
A 2½-year-old female child, first born of a non-consanguineous marriage, presented with complaints of excessive dryness and scaling of skin, present since birth. There was a history of collodion membrane, shedding of which was followed by redness and scaling all over the body. No bullous lesions or erosions were noted. A history of exacerbation of scaling in summers was present. At around 6 months of age the child developed abdominal distension. There was no history of associated fever, vomiting, difficulty in feeding, jaundice, dark-colored urine, or clay-colored stools. There was not a history of seizures, gait abnormalities, muscle weakness, deafness, or visual impairment. There was no history of developmental delay.
The younger female sibling, 1-month-old at the time of presentation, was also afflicted with a similar skin disorder. There was no history of collodion membrane or any history suggestive of systemic involvement in the younger sibling. There was no history of a similar disorder in any of the other family members.
 |  |
| Figure 1 | Figure 2 |
|---|
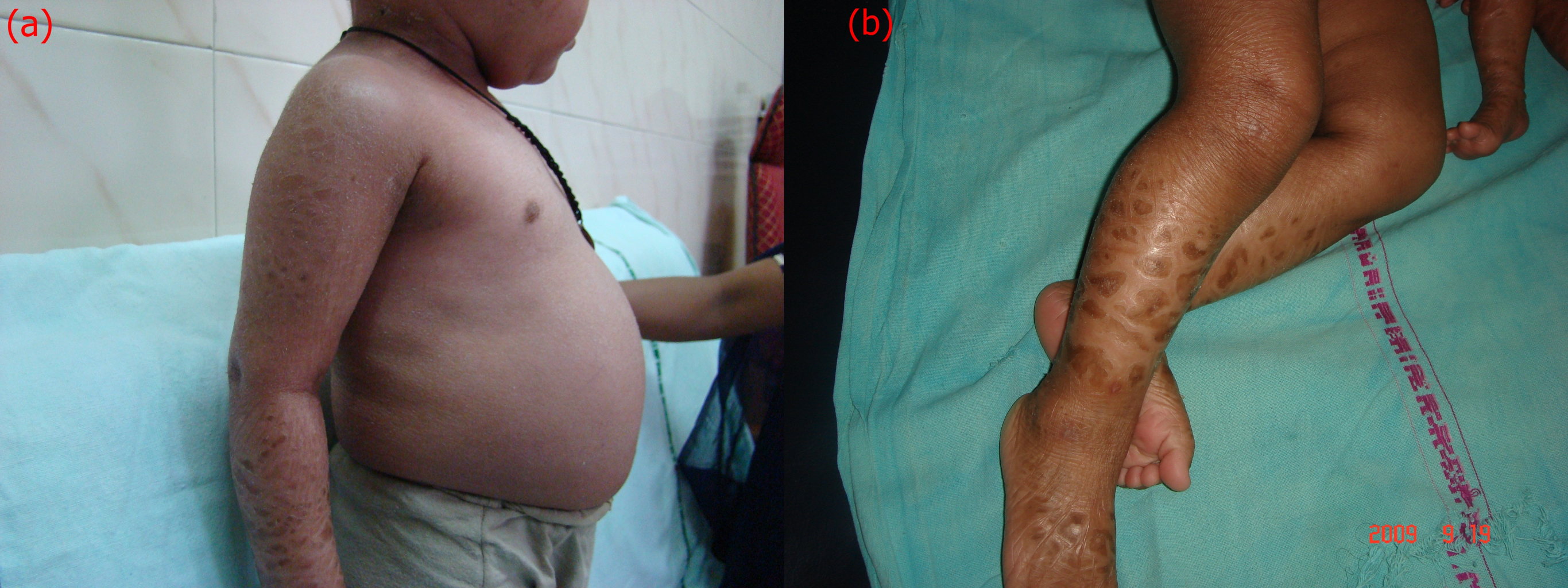
Examination of the older sibling revealed generalized scaling with mild erythema. The scales were fine, whitish, and semi-adherent over the trunk and larger, brownish, polygonal, and firmly adherent over the extremities and side of the face (Figures 1a and 1b). Scalp and flexures were also involved and fissuring was present in the retro-auricular area. Palms and soles showed hyperlinearity. There was no evidence of keratosis pilaris. Hair, teeth, nails, and mucosae appeared normal on examination. In addition, there was pallor and enlargement of the cervical and left axillary group of lymph nodes. Abdominal distension was also present and palpation revealed hepatomegaly. The liver was enlarged 5.5 cm below the right costal margin. It was firm and non-tender with a smooth outline. Muscle tone and reflexes were normal. Her general physical appearance exhibited mild wasting and stunting. Ophthalmological evaluation revealed bilateral lower lid ectropion (Figure 2a) and mild conjunctival congestion in both the eyes. ENT evaluation was normal and there was no evidence of developmental delay.
Examination of the younger sibling revealed generalized erythema along with diffuse scaling in a pattern and morphology similar to that of her elder sibling (Figure 2b). Scalp involvement in the form of scaling and patchy alopecia was also present. Systemic examination was within normal limits and ophthalmological and ENT evaluation showed no visual or auditory deficit.
Investigations
 |
| Figure 3 |
|---|
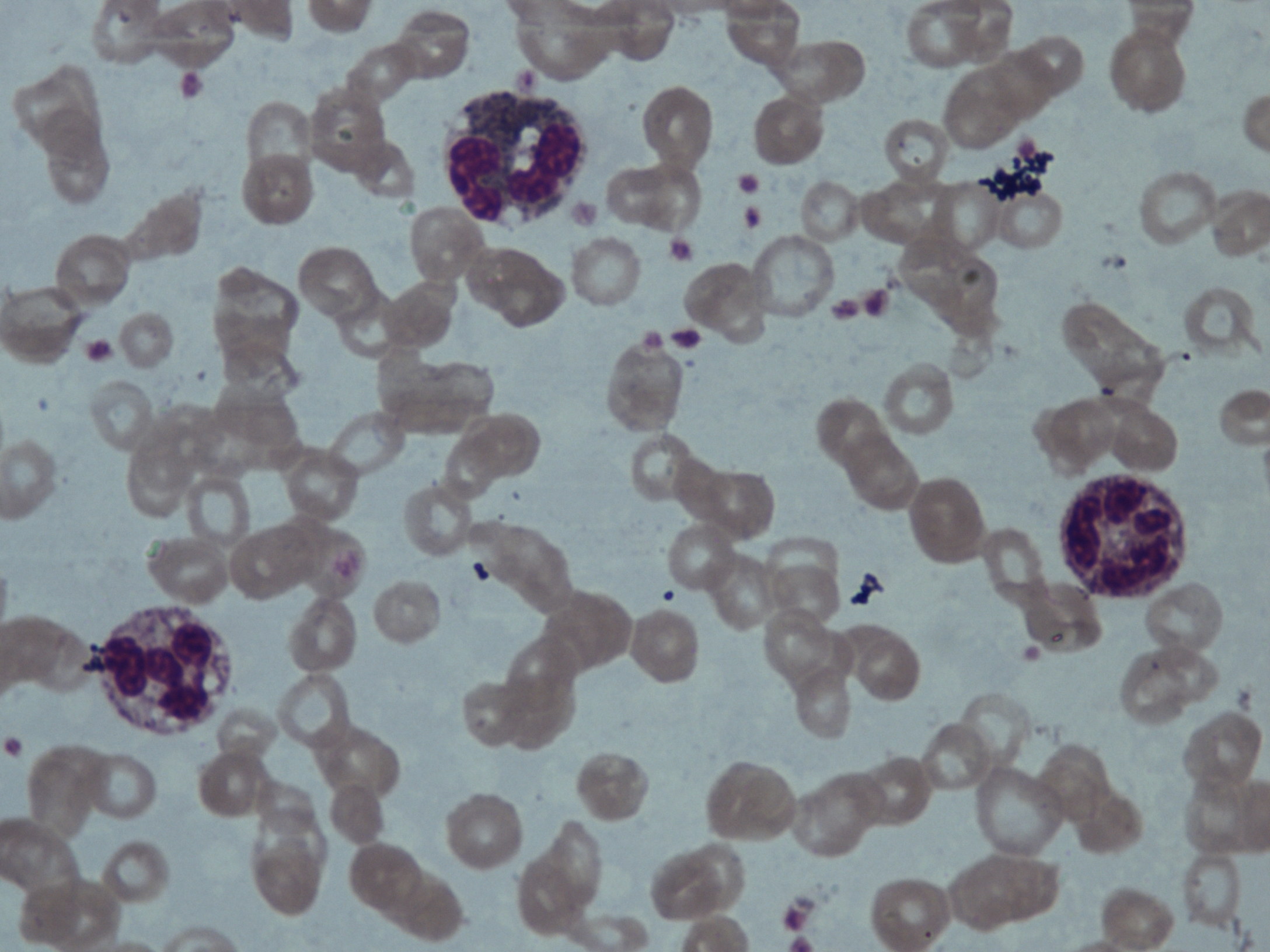
The older sibling was anemic with a hemoglobin of 6.7 gm. Peripheral smear showed microcytic hypochromic anemia along with cytoplasmic vacuoles in granulocytes and monocytes that stained positive for Sudan Black (Figure 3). Serum biochemistry revealed abnormal liver function tests (AST 69 IU/L, ALT 53IU/L). The lipid profile was also abnormal with a triglyceride level of 260mg/dl. Muscle-derived enzymes were elevated with a total creatinine kinase (CK) of 140 IU/L, CK-MB 35 IU/L, and LDH 903 IU/L. Jordan Anomaly could be demonstrated in the leukocytes of the younger sibling also. Her liver function tests and CK values were also elevated (AST-98 IU/L, ALT-52 IU/L, CK-58 IU/L, CK-MB 26 IU/L, and LDH-731 IU/L) but triglyceride levels were normal. Ultrasonography of the abdomen in the elder sibling showed the liver to be 12 cm (enlarged) and homogenous with altered echotexture, suggestive of fatty change in the older child. Ultrasonography was normal in the younger child. ECG was normal in both of the patients.
Skin biopsy revealed hyperkeratosis with hypogranulosis, increased vacuolation of the basal layer, and a mild perivascular dermal infiltrate. Fine needle aspirate of the lymph nodes revealed reactive lymphadenitis.
A diagnosis of Dorfman Chanarin syndrome was made in both the siblings based on the peripheral smear finding of Jordan anomaly along with NBIE. Screening of the family members including the parents and maternal and paternal grandmothers for Jordan anomaly was negative.
Discussion
Dorfman Chanarin syndrome usually manifests as congenital ichthyosiform erythroderma, but reports of patients with lamellar ichthyosis and erythrokeratoderma variabilis-like pictures also exist [10, 11]. Ectropion, eclabium, flexural lichenification, and palmoplantar hyperkeratosis are other associated cutaneous findings. The liver is affected in nearly 64 percent of cases and mostly manifests as hepatomegaly [12]. The most common pathology observed is steatohepatitis, which may later progress to fibrosis and cirrhosis. Myopathies are seen in around 60 percent of the cases [13]. In most of the affected patients muscle involvement goes unnoticed, but it may manifest as slowly progressive weakness of proximal limb muscles. Neurologic abnormalities include mental retardation, seizures, gait abnormalities, microcephaly, and sensorineural deafness. Other reported associations include growth retardation, cataracts, ptosis, nystagmus, cardiomyopathy, splenomegaly, intestinal anomalies, and short stature. Rickets and renal involvement have also been recently reported with DCS [14, 15].
Both the siblings in our report had NBIE as the presenting feature. In addition, the younger sibling also had patchy alopecia, which is rarely described as part of this entity. Liver involvement in the form of elevated liver enzymes in both and hepatomegaly in the elder sibling were clues to the correct diagnosis and caused us to examine the peripheral blood smear for Jordan anomaly. The triglyceride levels were elevated in the elder sibling and towards the higher range of normal in the younger one. Previous reports of serum lipid profiles in patients have been inconsistent with only a few cases in which abnormal lipid levels were mentioned in the literature [5, 16]. The muscle-derived enzymes were also markedly increased, but EMG and muscle biopsy could not be carried out; the parents refused to give consent. Although both of the offspring were affected, the parents denied any history of consanguinity. Neither the parents nor other family members examined showed any evidence of ichthyosis or other features of this disorder. Screening of the peripheral smear for vacuolization in leukocytes was also unsuccessful. Some reports suggest examination of eosinophils as a useful aid for detecting carriers. However, the general low concentration of eosinophils in a normal blood smear (2-4%) and the fact that not all eosinophils may show the characteristic vacuolation could lead to false negative results [17].
Diagnosing DCS is fairly simple and the importance of screening the peripheral blood smear for Jordan anomaly in all cases of congenital ichthyosis has been emphasized repeatedly by various authors. However, the procedure for investigating the patient to rule out any systemic involvement has not been previously discussed thoroughly. Table 1 lists the battery of investigations to be performed in patients with DCS. This listing should be of benefit to clinicians in the work up of this rare metabolic disorder.
Emollients remain the mainstay of treatment. Abnormal liver function in most of the patients calls for extreme caution before starting oral retinoids in cases with severe ichthyosis. Dietary supplementation of eicosapentanoic acid and docoahexanoic acid combined with the use of emollients was found to improve erythema and scaling in a 52-year-old with DCS [18]. Low protein, low fat, and high carbohydrate regimens are reported to be beneficial. Ursodeoxycholic acid and vitamin E have been recommended in steatohepatitis because of their cytoprotective and antioxidant effects. The role of dietary modification remains controversial and a recent report found no improvement in the ichthyosis or transaminase levels after 5 months of follow up [12]. Our patients are using emollients along with dietary intervention and are being followed up regularly.
The peculiarities in our case include the presence of this disorder in both female siblings along with alopecia in the younger sibling. Hyperlipidemia, noted in one of our patients, is also not a common association. A high index of suspicion may lead to higher rates of detection of this rare disorder. Like other authors, we too re-emphasize the need for screening the peripheral blood smear for leukocyte vacuolization in all cases of congenital ichthyosis.
References
1. Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gastroenterol Nutr. 1997 Nov;25(5):541-7. [PubMed]2. Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordan's Anomaly in White Blood Cells: Report of Case. Blood. 1966 Aug;28(2):258-65. [PubMed]
3. Pahwa M, Kar R, Singh A, Goel A, Ramesh V, Jain R. Chanarin-Dorfman syndrome with eccrine gland vacuolation: a case report. Int J Dermatol. 2008 Dec;47(12):1257-9. [PubMed]
4. Nanda A, Sharma R, Kanwar AJ, Kaur S, Dash S. Dorfman-Chanarin syndrome. Int J Dermatol. 1990 Jun;29(5):349-51. [PubMed]
5. Tullu MS, Muranjan MN, Save SU, Deshmukh CT, Khubchandani SR, Bharucha BA. Dorfman-Chanarin syndrome: A rare neutral lipid storage disease. Indian Pediatr. 2000 Jan;37(1):88-93. [PubMed]
6. Gupta P, Kaur G. Chanarin Dorfman syndrome neonatal diagnosis and 3-year follow-up. Indian Pediatr. 2005 Oct;42(10):1054-5. [PubMed]
7. Gandhi V, Aggarwal P, Dhawan J, Singh UR, Bhattacharya SN. Dorfman-Chanarin syndrome. Indian J Dermatol Venereol Leprol. 2007 Jan-Feb;73(1):36-9. [PubMed]
8. Lefèvre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, Ozgüc M, Lathrop M, Prud'homme JF, Fischer J. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001 Nov;69(5):1002-12. [PubMed]
9. Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 2006 May;3(5):309-19. [PubMed]
10. Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral lipid storage disease: A new disorder of lipid metabolism. Br Med J. 1975 Mar 8;1(5957):553-5. [PubMed]
11. Pujol RM, Gilaberte M, Toll A, Florensa L, Lloreta J, González-Enseñat MA, Fischer J, Azon A. Erythrokeratoderma variabilis-like ichthyosis in Chanarin-Dorfman syndrome. Br J Dermatol. 2005 Oct;153(4):838-41. [PubMed]
12. Selimoglu MA, Esrefoglu M, Gul M, Gungor S, Yildirim C, Seyhan M. Chanarin-Dorfman syndrome: Clinical features of a rare Lipid Metabolism Disorder. Pediatr Dermatol. 2009 Jan-Feb;26(1):40-3. [PubMed]
13. Brunoa C, Dimauro S. Lipid storage myopathies. Curr Opin Neurol. 2008 Oct;21(5):601-6. [PubMed]
14. Taskin E, Akarsu S, Aygun AD, Ozlu F, Kilic M. Rickets with Dorfman-Chanarin Syndrome. Acta Haematol. 2007;117(1):16-9. [PubMed]
15. Aksu G, Kalkan Ucar S, Bulut Y, Aydinok Y, Sen S, Anal O, Simsek Gosen D, Darcan S, Coker M, Kutukculer N. Renal Involvement as a Rare Complication of Dorfman-Chanarin Syndrome: A Case Report. Pediatr Dermatol. 2008 May-Jun;25(3):326-31. [PubMed]
16. EI-Kabbany Z, Elsayed SM, Rashad M, Tareef R, Galal N. Dorfman-Chanarin syndrome in Egypt. Am J Med Genet A. 2003 Aug 15;121A(1):75-8. [PubMed]
17. Wollenberg A, Geiger E, Schaller M, Wolff H. Dorfman-Chanarin syndrome in a Turkish kindred: Conductor diagnosis requires analysis of multiple eosinophils. Acta Derm Venereol. 2000 Jan-Feb;80(1):39-43. [PubMed]
18. Bañuls J, Betlloch I, Botella R, Sevila A, Morell A, Román P. Dorfman-Chanarin syndrome (neutral lipid storage disease). A case report. Clin Exp Dermatol. 1994 Sep;19(5):434-7. [PubMed]
© 2011 Dermatology Online Journal