Congenital tufted angioma: Case report and review of the literature
Published Web Location
https://doi.org/10.5070/D39103z6s5Main Content
Congenital tufted angioma: Case report and review of the literature
Fernando Toledo Alberola, Isabel Betlloch MAS MD, Laura Cuesta Montero MD, Irene Ballester Nortes MD, Nuria Latorre Martínez
MD, Almudena Monteagudo Paz MD
Dermatology Online Journal 16 (5): 2
Dermatología, Hospital General Universitario De Alicante, Alicante, Spain. fernandotoledoalberola@hotmail.comAbstract
Tufted angiomas (TA) are rare benign vascular tumors of unknown pathogenesis. Most appear during childhood; approximately 25 percent are congenital and 50 percent appear in the first year of life. According to the literature, TA that are present at birth or in the first year of life have a greater tendency to spontaneously regress than do those that appear later in life. Their clinical presentation is non-specific and characterized by bluish-erythmatous plaques or nodules. The differential diagnosis includes infantile hemangiomas, congenital hemangiomas, kaposiform hemangioendothelioma and vascular malformations. Tufted angiomas have a characteristic histology consisting of a proliferation of endothelial cells forming lobules with the typical “shotgun” distribution. We report a case of congenital TA and review the cases of congenital TA described to date in the literature in order to highlight the different characteristics of congenital and acquired TA.
Introduction
A tufted angioma (TA), first described by Nakagawa in 1949 as an angioblastoma, is a rare benign vascular tumor, which appears to be more common in the Asian population. However, numerous cases have been reported in the USA and Europe [1].
Other names such as juvenile TA or progressive capillary hemangioma have been given to this clinical entity and some authors have even questioned whether Nakagawa's angioblastoma and TA are in fact the same entity [2].
Although most cases appear in childhood, about 25 percent are congenital and 50 percent appear in the first year of life [2]. Cases in which the tumors appear later in adults, in association with pregnancy [3], and in recipients of solid organ transplant [4] have also been described. Most TA are sporadic, but cases have also been described in which they were hereditary with an autosomal dominant pattern [5].
According to the literature, TA that present at birth or in the first year of life have a greater tendency to spontaneously regress than do those acquired later in life [2]. We present a case of congenital TA and review the literature on cases of TA described to date.
Case report
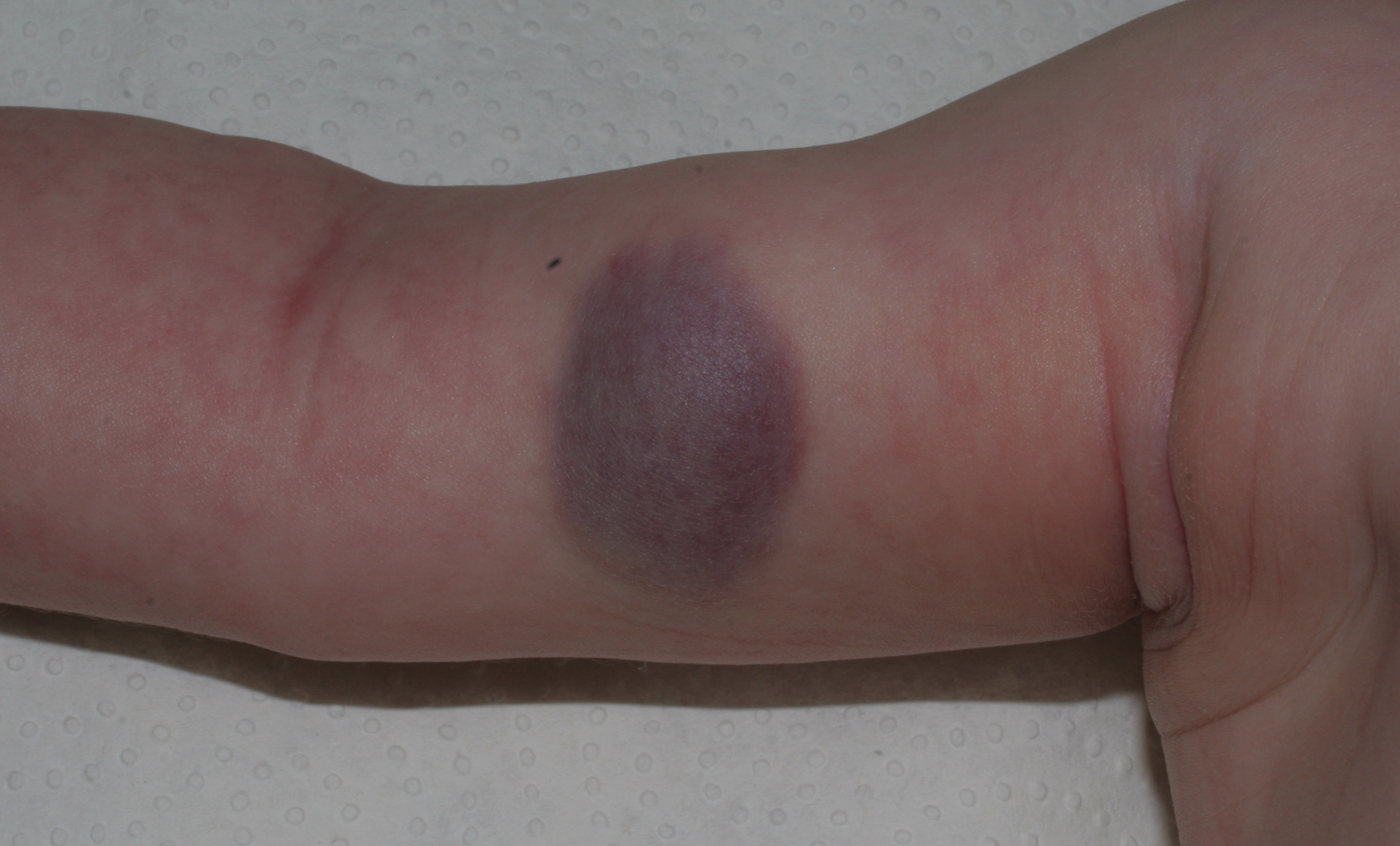 |
| Figure 1 |
|---|
| Figure 1. Soft, raised, purple plaque measuring 4 x 2 cm, surrounded by a pale peripheral halo, on the medial aspect of the right arm |
A 2-month-old baby girl presented with a stable, asymptomatic tumor, present since birth on the medial aspect of her right arm.
On physical examination, there was a soft, non-pulsatile, raised purplish plaque, measuring 4 x 2 cm, exhibiting no change in temperature, and surrounded by a pale peripheral halo (Figure 1).
The differential diagnosis between infantile hemangioma, non-involuting congenital hemangioma (NICH), rapidly involuting congenital hemangioma (RICH), and other vascular neoplasias was considered (Table 1).
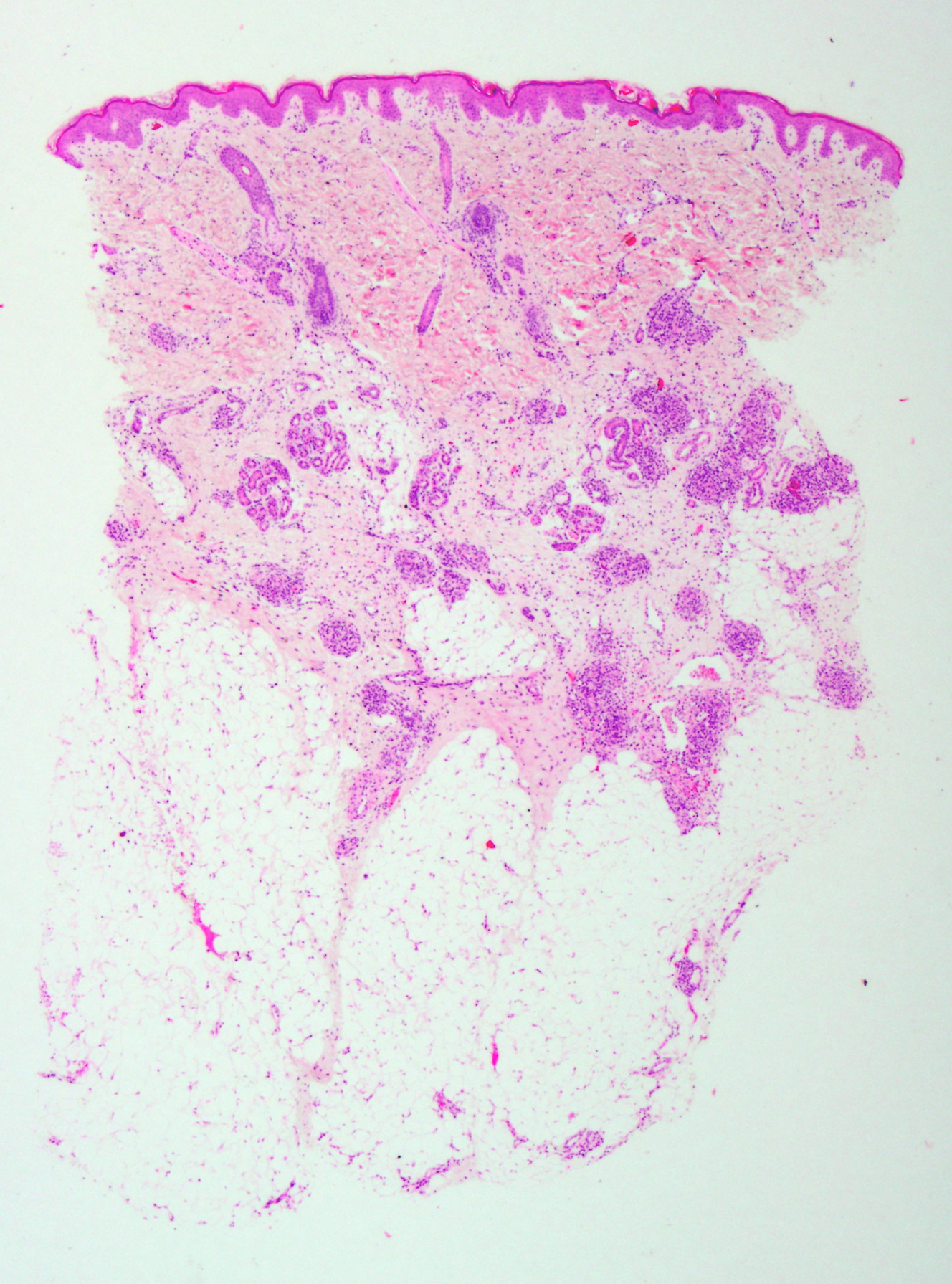 |  |
| Figure 2 | Figure 3 |
|---|---|
| Figure 2. Capillary clusters in dermis and subcutaneous cell tissue (H&E, x2) Figure 3. Glomeruloid lobules, made up of fusiform endothelial cells, without atypia and mitosis (H&E, x20) | |
On skin biopsy, capillary clusters were seen in the form of glomeruloid lobules, consisting of fusiform endothelial cells around the vascular plexus of the dermis and subcutaneous cell tissue. The proliferations were non-atypical and showed no mitoses (Figures 2 and 3). The tumor showed inmunopositivity for CD31, CD34, and alpha-actin (Figure 4). Immunohistochemistry for GLUT-1 and factor VIII was negative (Figure 4).
Results of the blood analysis and coagulation study showed no abnormalities. On Doppler echography, a high flow vascular lesion was seen.
 | 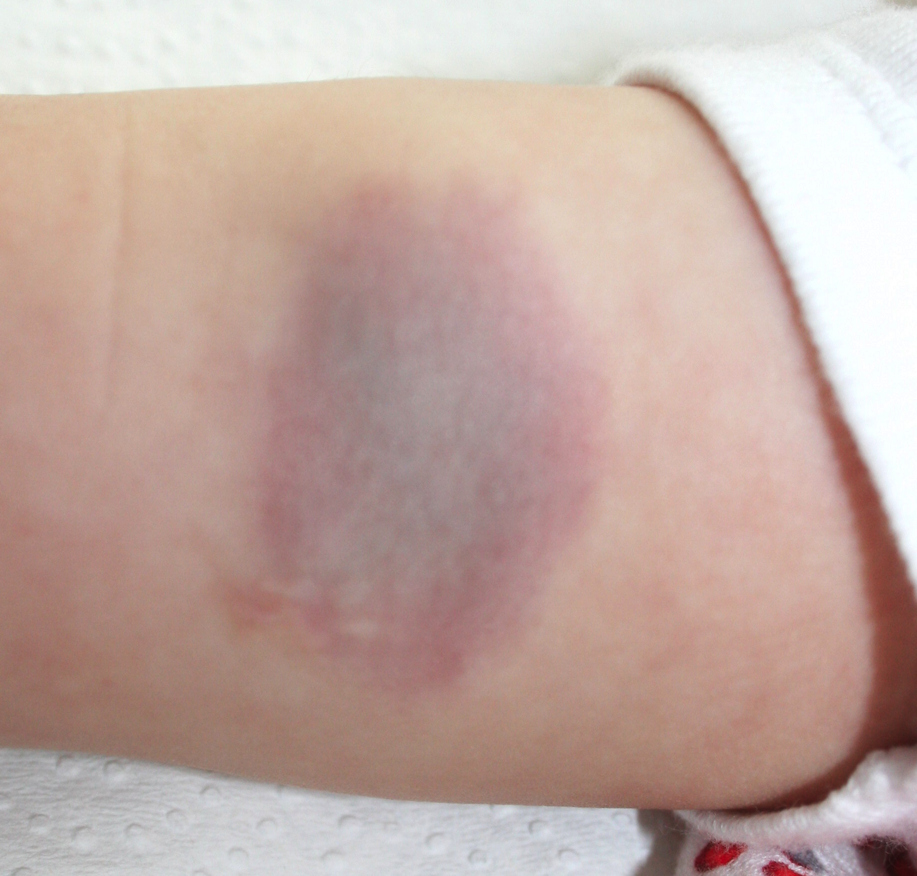 |
| Figure 4 | Figure 5 |
|---|---|
| Figure 4. Immunostaining for CD31, CD34 and alpha-actin was positive, with factor VIII negative (Original magnification: x2) Figure 5. Clinical image six months after presentation. A reduction in the volume of the lesion and a change in its color may be seen. | |
The diagnosis of congenital TA was reached and, given the tendency of this type of angioma to regress, it was decided to simply keep the patient under observation; the parents were informed that the tumor would probably resolve spontaneously.
After a 6-month follow up a reduction in volume and change in color had occurred, indicating that the tumor is in the process of regression (Figure 5).
Results
We reviewed all the cases of congenital TA described to date in the literature (only lesions present at birth), analyzing patient characteristics including sex, clinical manifestations, site, course and complications in each of the 27 cases found [6-16] (Table 2).
We found that as regards both congenital and acquired TA, there was no predominant gender (9 ♀ / 9 ♂, in the 18 cases in which the gender was specified by the authors).
Regarding the clinical picture, TA usually present as bluish-erythematous plaques or nodules of varying size, located preferentially on the arms and legs, which coincides with the clinical picture in our patient. Likewise, acquired TA usually present as bluish or reddish indurated plaques or nodules, measuring 2 to 5 cm, located preferentially on the trunk, neck and arms [13, 14].
In general TA may be associated with hyperhydrosis and hypertricosis [2]. Recently, cases with unusual clinical presentations have been described, such as TA with spontaneous regression [6], metastatic forms [17], forms mimicking cavernous hemangioma [10]or tumors located on top of pre-existing port-wine stains [18].
In our case, the clinical presentation led us to consider the possibility that it was a congenital hemangioma (NICH or RICH), vascular malformation, or kaposiform hemangioendothelioma.
The typically non-specific clinical features necessitate that the tumor be diagnosed histologically. In both the congenital and acquired form, a proliferation may be seen at the level of the dermis, consisting of clusters of a dual population of immature endothelial cells and pericytes, with a characteristic shotgun-like pattern. Each lobule is made up of non-atypical fusiform endothelial cells with no mitoses. These are arranged around the vascular plexus of the dermis and subcutaneous cell tissue, into which they may protrude with a half-moon distribution. In contrast to infantile hemangiomas, they always have negative GLUT-1 immunohistochemistry [14, 19, 20].
The pathogenesis of TA is not known but is likely related to increased secretion of growth factors such as IL-8, which may influence angiogenesis, favoring development of numerous vascular clusters [21].
With regard to their course, acquired TA may exhibit a slow growth phase for months or years, with little tendency to spontaneously regress (15.9% of cases [9]). In congenital or early TA, the prognosis appears to be different. There is a greater tendency to involute, which usually occurs in 95 percent of cases within 6 months to 2 years [15].
These findings coincide with those found in our review of the literature. Of the 27 cases described, including our own, the course was known in 16. In 15 of the cases (93%), there was partial or complete regression of the lesion, which occurred between the ages of 4 months and 6 years. Only in one of the cases reported [14] did the lesion remain stable over time and persisted after a 7-year follow-up. These data support the theory that congenital TA have a different course compared to that of other TA. Congenital TA have a greater tendency to regress, thereby confirming that the best therapeutic option in such cases is to keep the patients under observation.
The treatment recommended in cases in which vital organs are compromised, when symptoms are severe, or for aesthetic reasons is surgical excision [21]. However, there are other therapeutic options available, but high doses of systemic corticosteroids, vincristine, and other options have given inconsistent results [22]. Perhaps the recently introduced selective β blockers for the treatment of certain cases of infantile hemangiomas [23] would be a valid, adequate therapeutic option for the treatment of cases of TA with indications for aggressive treatment.
One of the most serious and potentially life-threatening complications described in TA is the Kasabach-Merritt syndrome [24] or consumption coagulopathy. In this case, treatment is usually aggressive with coagulation factors and transfusions of platelets, systemic corticosteroids, embolization, surgical removal, chemotherapy, or radiotherapy. However, in congenital TA, only one case of Kasabach-Merritt syndrome has been reported, and this responded very well to treatment with vincristine [8].
Conclusions
We report a case of congenital TA, a rare benign vascular neoplasm with a great tendency to spontaneously regress [15] and little likelihood of malignant transformation. In the absence of complications, the best therapeutic option is to keep the patient under observation.
The clinical presentation of congenital TA is similar to that of acquired TA. They share the same epidemiological and histological characteristics and therapeutic options but they differ in that congenital TA are preferentially located on the limbs, the rate of complications is lower, and the prognosis is better.
References
1. Wilson-Jones E. Malignant vascular tumors. Clin Exp Dermatol. 1976; 1: 287-312. [PubMed]2. Okada E, Tamura A, Ishikawa O, Miyachi Y. Tufted angioma (angioblastoma): case report and review of 41 cases in the Japanese literature. Clin Exp Dermatol. 2000; 25: 627-630. [PubMed]
3. Kim YK, Kim HJ, Lee CG. Acquired tufted angioma associated with pregnancy. Clin Exp Dermatol. 1992; 17: 458. [PubMed]
4. Chu P, LeBoit PE. An eruptive vascular proliferation resembling acquired tufted angioma in the recipient of a liver transplant. J Am Acad Dermatol. 1992; 26: 322. [PubMed]
5. Heagerty AH, Rubin A, Robinson TW. Familial tufted angioma. Clin Exp Dermatol. 1992; 17: 344. [PubMed]
6. Lam WY, Lai FMM, Look CN, Choi PC, Allen PW. Tufted angioma with complete regression. J Cutan Pathol. 1994; 21: 461-466. [PubMed]
7. Jang KA, Choi JH, Sung KJ, et al. Congenital linear tufted angioma with spontaneous regression. Br J Dermatol. 1998; 138: 912-914. [PubMed]
8. Enjolras O, Wassef M, Dosquet C, et al. Syndrome de Kasabach-Merritt sur angiome en touffes congenital. Ann Dermatol Venereol. 1998: 125: 257-60. [PubMed]
9. Igarashi M, Oh-i T, Koga M. The relationship between angioblastoma (Nakagawa) and tufted angioma: report of four cases with angioblastoma and a literature-based comparision of the two conditions. J Dermatol. 2000; 27: 537-542. [PubMed]
10. Kim KJ, Lee MW, Choi JH, Sung KJ, Moon KC, Koh JK. A case of congenital tufted angioma mimicking cavernous hemangioma. J Dermatol. 2001; 28: 514-415. [PubMed]
11. Kimura Y, Hata M, Kawana S, Itoh F: Angioblastoma (Nakagawa) showing spontaneous regression (in Japanese). Pract Dermatol 2000; 22: 53-56.
12. Kato F, Kishi H, Takahashi H, Horikoshi T. A case of a angioblastoma (Nakagawa) which disappeared spontaneously (in Japanese). Rinsho Hifuka 2000; 54: 173-175.
13. Wong SN, Tay YK. Tufted angioma: a report of five cases. Pediatr Dermatol. 2002; 19: 388-393. [PubMed]
14. Satter EK, Graham BS, Gibas NF. Congenital tufted angioma. Pediatr Dermatol. 2002; 19: 445-7. [PubMed]
15. Browning J, Frieden I, Baselga E, Wagner A, Metro D. Congenital, self-regressing tufted angioma. Arch Dermatol. 2006; 142: 749-751. [PubMed]
16. Barco D, Baselga E, Ribé A, Alomar A. Angioma en penacho congénito. Actas Dermosifiliorg. 2008; 99: 423-24.
17. Bang RH, Padilla RS. Metastatic tufted angioma. Int J STD AIDS. 2000; 11: 414. [PubMed]
18. Kim TH, Choi EH, Ahn SK, Lee SH. Vascular tumors arising in port-wine stains: two cases of pyogenic granuloma and a case of acquired tufted angioma. J Dermatol. 1999; 26: 813-6. [PubMed]
19. North PE, Waner M, Mizeracki A, Mihm MC Jr. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol 2000; 31: 11-22. [PubMed]
20. Requena L, Sangueza OP. Cutaneous vascular proliferation. Part II. Hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997; 37: 887-919. [PubMed]
21. Bernstein EF, Cantor G, Howe N, Savit RM, Koblenzer PJ, Uitto J. Tufted angioma of the thigh. J Am Acad Dermatol. 1994; 31: 307-311. [PubMed]
22. Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005; 44:1045-7. [PubMed]
23. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008; 358: 2649-51. [PubMed]
24. Enjolras O, Wassef M, Mazoyer E, Frieden IJ, Rieu PN, Drouet, et al. Infants with Kasabach-Merritt syndrome do not have “true” hemangiomas. J Pediatr. 1997; 130: 631-40. [PubMed]
© 2010 Dermatology Online Journal