Human skin keratinocytes modified by a Friend-derived retroviral vector: A functional approach
Published Web Location
https://doi.org/10.5070/D37xb8v9gzMain Content
Human skin keratinocytes modified by a Friend-derived retroviral vector: A functional approach
M Arango1, C Chamorro1, O Cohen-Haguenauer2, M Rojas3, and LM Restrepo1
Dermatology Online Journal 11 (2): 2
1. Grupo de Ingeniería de tejidos y terapias celulares. Facultad de Medicina Universidad de Antioquia Medellín, Colombia.
lrestre@catios.udea.edu.co2. Eurogenethy and Laboratoire de Biotechnologie and pharmacogie genetique appliquées (LBPA). Ecole
Normale superieure de Chachan. odile.haguenauer@lbpa.ens-chachan.fr 3. Department of Immunology and Infectious Diseases, Harvard
School of Public Health. Boston USA. mrojasl@epm.net.co
Abstract
The goal of this study was to test the efficiency and possible functional effects of a Friend Leukemia derived retrovirus vector (FOCH29-NeoR) on cultured human keratinocytes, obtained from skin biopsy samples. The keratinocytes were grown and infected with filtered Friend vector supernatant. After one or two doses of infection, one duplicate of the culture was submitted to selection with G418; the other one was utilized for DNA extraction and PCR modification detection. Transduction efficiency was 46.66 percent and 47.22 percent for one and two doses of infection respectively (range 100 to 15 %). Colony Forming Efficiency (CFE) assays were done with Rodhamine-B staining in nonselected modified cultures and negative controls. There was no difference in CFE (% CFE= 10.74±6.53 negative control vs % CFE= 9.22±5.45 with one dose, and % CFE=10.03±5.74 with two doses of infection). Nevertheless, the cell-cycle analysis done by Propidium Iodade (PI) incorporation and colchicine-arrest assays in nonselected transduced and nontransduced cells show that transduced keratinocytes have a longer time to enter G2. As far as we know, this is the first report of retroviral transduction-induced changes in the cell cycle done on human keratinocytes. This observation is very important because retroviral vectors of genes, such as platelet derived growth factor (PDGF) or vascular endothelial growth factor (VEGF), are expected to facilitate the implementation of these modified cultures for tissue grafting and skin substitute development and potentiate the effectiveness of the grafts.
Introduction
Skin is an attractive tissue for the development of gene therapy for skin and hair treatment and systemic genetic disorders [1]. Some characteristics of the keratinocytes have made these cells ideal targets for gene transfer. They can be expanded in vitro, their culture conditions have been well established, and they are accessible and easy to monitor after transduction [2]. Keratinocytes have a wide spectrum of clinical applications because they produce different growth factors, cytokines, and secreted proteins with local and systemic action.
Phenotypic reversion has been achieved in vivo for junctional epidermolysis bullosa and lamellar ichthyosis and in vitro for xeroderma pigmentosum [3, 4, 5]. These results should prompt clinical trials based on transplantation of artificial epithelia reconstructed ex vivo using genetically modified keratinocytes. Promising results have also been obtained in metabolic diseases [6].
Biological, physical and chemical systems have been used to modify murine and human keratinocytes [2]. From these, Moloney murine-leukemia virus-derived retroviral vectors are the most utilized vectors in preclinical and clinical protocols [4, 5, 7]. Even though these vectors have been the most extensively studied and well characterized, problems related to inactivation of gene expression by cytokines such as IFN-γ [8] and host immune responses have been reported for vectorized transgenes [9, 10]. A persistent problem for keratinocyte gene therapy with retroivurs vectors has also been the transient nature of transgene expression in vivo [11, 12].
Recently, it has been demonstrated that a transgene expressing EGFP from liposome transfected cultures of epidermal keratinocytes undergoes cell-cycle arrest and initiates terminal differentiation. This same study showed in contrast that transduction with a retroviral vector expressing the EGFP gene does not alter the potential growth of the keratinocytes. No decrease in CFE was detected with this kind of vector [13]. The importance of establishing parameters and specific conditions for each cellular target and for each encapsidation line in diverse studies has been proven [14]. Accordingly, in this study we evaluate the ability of a Friend murine leukemia-derived vector (FOCH29-NeoR) [15] to transduce human skin keratinocytes. We also tested the possible effects of this vector on cell cycle and the proliferative capacity of primary cultured keratinocytes. Comparative experiments on cell cycle effects were done on 3T3 NIH cells. Recent reports confirm the importance of evaluating retroviral vectors effects on target cells at a basic level [16].
Methods
CelI isolation and culture
After informed consent, human epidermal keratinocytes were obtained from foreskin biopsies and samples were washed with phosphate-buffered saline (PBS) (Gibco). After treatment with penicillin (100U/mL) and streptomycin (100 μg/mL) (Gibco) for 30 min, the skin was finely minced with surgical scissors, mixed with 25-50 ml of 0.025 percent Trypsin/EDTA solution, transferred into a 125 mL flask and stirred gently at 37°C. Every 30 min, the suspended cells were harvested. The resulting cell suspension was washed with Dulbecco’s modified Eagle’s medium (DMEM) (Gibco), supplemented with 10 percent fetal bovine serum (FBS) (Gibco), resuspended, and counted. The viability was determined by trypan blue dye exclusion. Fresh Trypsin–EDTA was added to the flask repeating this treatment three or four times. Isolated cells were plated in serum free medium (KGM Clonetics).
Packaging cell line culture and supernatant production
The ϕ Crip FOCH29-NeoR [15] and the PG13-GFP [17] packaging cells were grown in DMEM high glucose (Gibco) and 10 percent FBS (Hyclone), penicillin-streptomycin 1 percent (Gibco) and L-glutamyne 2mM (Gibco) on 75 cm2 culture flasks, at 37°C, 5 percent CO2. Once confluent, 10 ml of fresh medium was added. After 24 hours the supernant was recovered and filtered with cellulose acetate filters 0.45 μm (Advantec MFS, INC). The titer of the FOCH29-NeoR packaging cell line supernatant measured on 3T3 NIH cells, oscillated between 4x106 ± 0 (n=3) and 3x107 ± 2.8x106 (n=7) colony-forming units/mL (cfu/mL). PG13-GFP titer was as reported [17].
Keratinocyte transduction
Keratinocytes were plated at 1x105 cells/well in serum free medium (KGM Clonetics). Cells were fed 3 times per week and incubated at 37°C in a humidified atmosphere with 5 percent CO2. At about 40-50 percent confluence, the cells were infected with 1 mL of viral supernatant in the presence of Polybrene (8 μg/mL Sigma) for one or two doses (6 hours between first and second dose). At 24 hours after transduction fresh medium was added. Duplicates including a negative control were performed for each condition.
Transduction evidence
At 48 hours after transduction, one culture duplicate was used for keratinocyte-transduced selection with medium containing G418 (400 μg/mL) for 14 days. The number of resistant colonies and the percentage culture confluence was determined microscopically.
The other duplicate was utilized for DNA extraction; when about 70 percent confluence was observed, the cells were trypsinated and lised with 100 μL of lisis Buffer (50mM KCl, 10mM Tris HCl pH 8.3, 25 mM MgCl2, 1mg/mL gelatin, 0.45 percent Np40, 0.45 percent Tween 20), plus 0.6 μL of Proteinase K (PK), 10 mg/ml, incubated for 1 hour at 55°C. PK was inactivated 10 min at 95°C; cell lysates were centrifuged for 2 minutes at 14000 rpm. Supernants containing DNA were stored at –20°C until use.
PCR
PCR was performed under the following conditions: DNA 5 μL, Taq polimerase (Gibco) 1 μL, dNTP (Gibco) 1 μL, PCR Buffer 1X (Gibco) 5 μL, MgCl2 (50mM Gibco) 0.7 μL and 1 μL primers, H2O was added for a final volume of 5O μL. The primers were as fallows: sense: GACGAGTTCTTCTGAGCGGG, antisense : GATCTGAACTTCTATTCTTG [18].
These primers amplified a 650 pb fragment. Amplification conditions were 1 cycle 5 min at 94°C, followed by 40 cycles of 30 min at 94°C, 20” at 55°C and 30 min at 72 °C for the final elongation time. PCR products were analyzed by 1-percent agarose-gel electrophoresis stained with ethidium bromide.
Determination of the colony formation efficiency (CFE)
A CFE assay was used to determine possible effects of transduction on the proliferative capacity of modified and non-modified keratinocytes. Non selected keratinocytes were used in these experiments after transduction. 3T3-Swiss mouse fibroblasts were irradiated (6000 rads) and plated (6x106) in a 100-mm culture dish. After 24 hours, 1x104 keratinocytes, modified for one or two doses, or not modified control cells were added; EGF (Austral Bilogicals) (10 ng/ml), was added in the first medium change. Cultures were maintained at 37°C, 5 percent CO2 from 11-12 days and fed 3 times weekly. After that time, culture medium was discarded and 2-ml Rodamine B was added for 5-10 min; then Rodamine B was discarded and washed with water. Percent CFE was calculated as follows: %CFE= (# Obtained colonies / 104 cells) X 100
The number and type of colonies obtained were determined microscopically; using previous reported criteria [19].
Cell cycle measurement by mitotic cell accumulation and propidium iodade (PI) staining.
One hundred thousand (1x105) nonselected transduced (by one or two doses supernatant) and nontransduced keratinocytes were plated in 2 mL KGM medium (six wells dish Nunc). After 5 days (60-70 % confluence), the cells were treated with colchicine (0.4 mg/mL) from 0 to 5 hours and then trypsinated. The cells were fixed (1x106) as described below for DNA staining with PI and then evaluated by flow cytometry. For Giemsa staining, medium was aspirated, cells were washed with PBS, and lysed with hypotonic solution for 3 minutes. They were also fixed with methanol:acetic acid (3:1) for 10 min followed by a 4 percent Giemsa stain for 5 min. Mitotic Index (MI= number of mitotic cells/total number of cells), was determined by counting at least 3000 cells (including mitotic and non mitotic cells for each condition).
The data obtained from MI was used for lineal regression to find cell cycle time, according to Puck and Steffen [20].
Cell cycle analysis by PI incorporation
The nonselected transduced keratinocytes and negative controls as well as 3T3 NIH murine fibroblasts were obtained after trypsine treatment. Cells were washed (1x106) and fixed with cold ethanol 70 percent. Then they were also washed 2-3 times with an isotonic buffer, and 500 μl PI/RNAase (30 U Sigma) was added. Cells were then incubated at 20°C for 30 min, washed with 2-ml PBS-KCl, resuspended in 500 μl PBS-KCL, and analyzed by flow cytometry (Coulter EPICS XL, Hialeah, Florida).
Data Analysis
Cytofluorometric data analyses were performed with Windows Multiple Document Interface (WinMD) version 2.8 (Scrips Resarch Institute, La Jolla, CA) “Stratgraphics Plus” versión 4, 1999 (Stratgraphics Corp Rockville, MD) and “Graphpad Prism” versión 3.0 (Graphpad Software Inc., San Diego).
Results
Keratinocyte transduction
The effect of G418 on human primary keratinocytes was determined first. After 14 days of selection 200 μg/mL as DL100 was found. Thus 400 μg/ml G418 were used to select retroviral transduced keratinocytes. The optimal degree of confluence to infect the keratinocyte cultures was established at 40-60 percent.
As shown in figure 1(B), 46.6 percent and 47 percent transduction efficiency were obtained with one or two doses of infection respectively, as detected by PCR (Fig. 1) and also evidenced by their resistance to G418 selection for 14 days. Transduction efficiency ranged from 100 to 15 percent. There was no difference between transduction with one dose or two doses of infection.
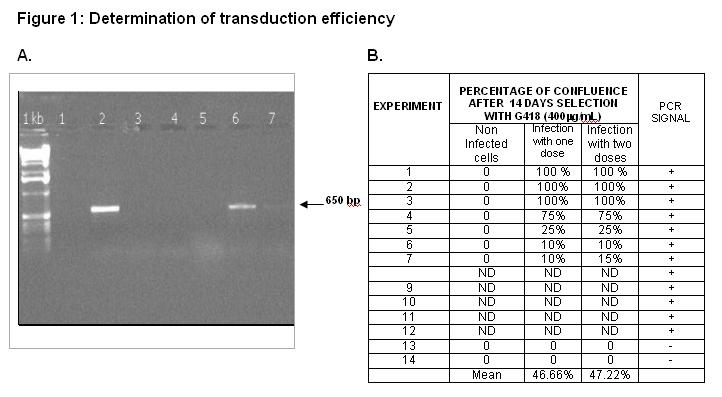{kind=link}
Colony Forming Efficiency (CFE)
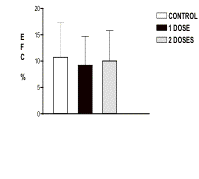
CFE tests were performed in cultures without selection from transduced and nontransduced keratinocytes. No significant statistical difference was observed between negative control (10.74 % ± 6.53) and one dose (9.22 % ±5.45) or two doses (10.03 % ± 5.74,) of infection (Fig. 2). These results suggest that the vector FOCH29-NeoR does not affect the capacity of keratinocytes to form colonies.
{kind=link}
Cell cycle analyses were tested by incorporating PI and blockade experiments with colchicine in keratinocytes and 3T3 NIH murine fibroblasts.
Keratinocytes
No significant differences were observed in the cellular distribution as determined by PI staining in the different phases of the cellular cycle (Fig. 3). On non selected transduced and non transduced cells, cell cycle blockade with colchicine was performed. Treatment with colchicine resulted in a time-dependent increase in the percentage of cells in G2/M in both transduced and nontransduced keratinocytes. However, with each hour in colchicine, a smaller percentage of transduced keratinocytes entering to G2/M was observed, compared to nontransduced (Fig. 4A). The slope of the function of mitotic accumulation obtained by PI incorporation in the transduced cells was significantly smaller (0.36 ±0.052, p<0.01) than for the non-transduced cells (1.48±0.15) (Fig. 4B).
{kind=link}
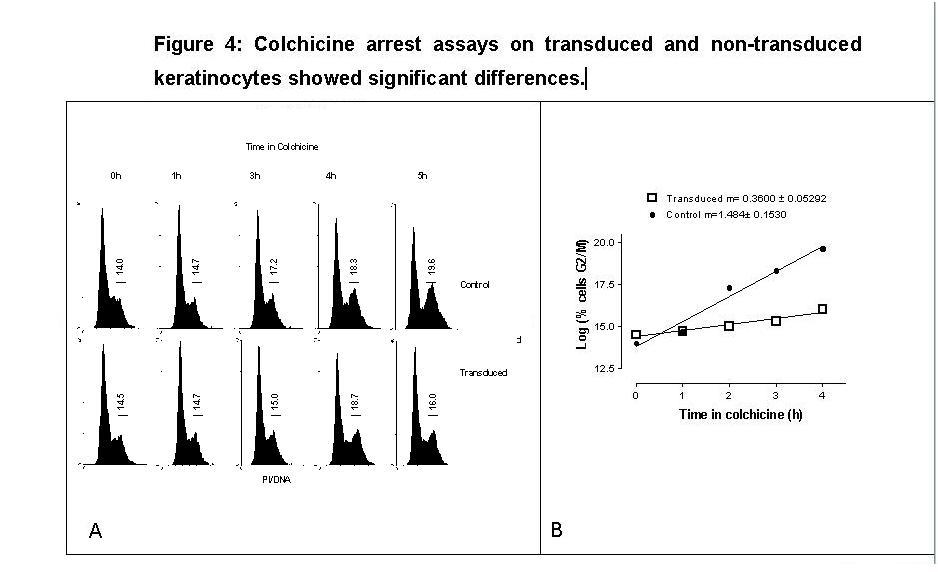{kind=link}
To further confirm these results, at different times in colchicine (0-5 hours) the cells were fixed in situ and stained with Giemsa. An increase in mitotic cells dependent on time was observed. At time 0, comparable mitotic indices were observed in modified and nonmodified keratinocytes. Thereafter, the mitotic index of the transduced cells was always lower than in nontransduced cells. The mitotic index was calculated by counting the number of mitotic cells in a total of 3000 cells. A linear regression of log (MI+1) was performed with time in colchicine. The values of the slope of transduced keratinocytes were significantly lower than for nontransduced cultures (0.0060±0.0007 and 0.0081±0.0060 respectively), which suggests that the transduced cells have a delay in the entrance to G2.
3T3 NIH murine fibroblasts
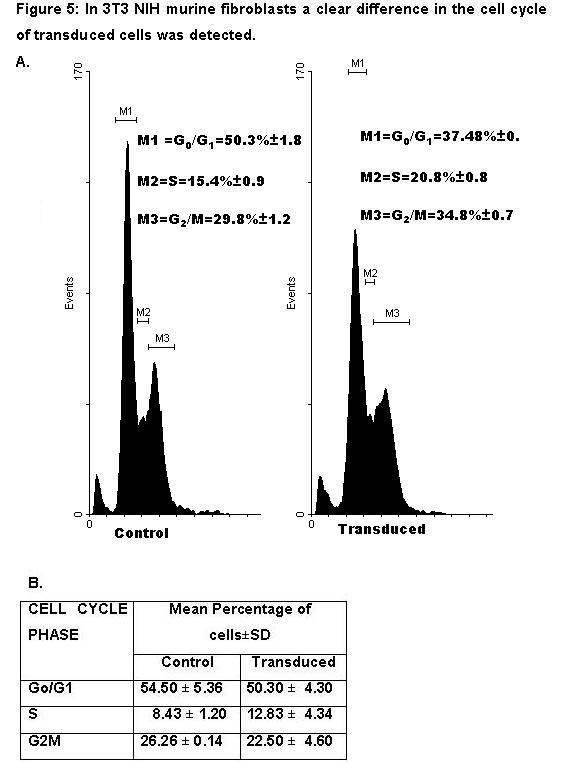
In order to determine whether the observed effect of transduction in keratinocytes was specific for these cells, control experiments were performed on the 3T3 NIH murine fibroblasts. A significant difference was found in cultures of 3T3 NIH transduced cells when there was a high percentage of cells in phases S (20.8 % ± 0.8) and G2/M (34.8 % ±0.7) compared to not transduced S (15.4 % ±0.9) and G2/M (29.8 % ±1.2) cultures (Fig. 5A). Fig. 5B shows the comparative results obtained with the PG13-GFP vector. No differences were observed in the percentage of cells in phases S(12.83±4.34) and G2/M (22.5±4.60) transduced cultures compared to not transduced S (8.43±3.36) and G2/M (26.26±0.14) cultures.
{kind=link}
Experiments of mitotic blockade were also carried out in 3T3 NIH transduced and nontransduced cells. There was a time dependent increase in the number of cells in G2/M. A smaller percentage of cells in G2/M was observed in non transduced cells (17 %) at the beginning of the blockade compared to the transduced cells (22.6 %); in spite of the small differences observed in the percentages of cells in G2/M even four hours after the blockade (38.5 % in not transduced and 39.3 % in transduced) (Fig. 6A), it was observed that the velocity of entrance to G2 of the modified cells was less than in the non modified cells which is reflected in the regression slope of log percent cells in G2/M vs. time, which was also significantly smaller in the transduced cells (0.1196 ± 0.02) than in the nontransduced cells (0.1491 ± 0.01), Fig. 6B).
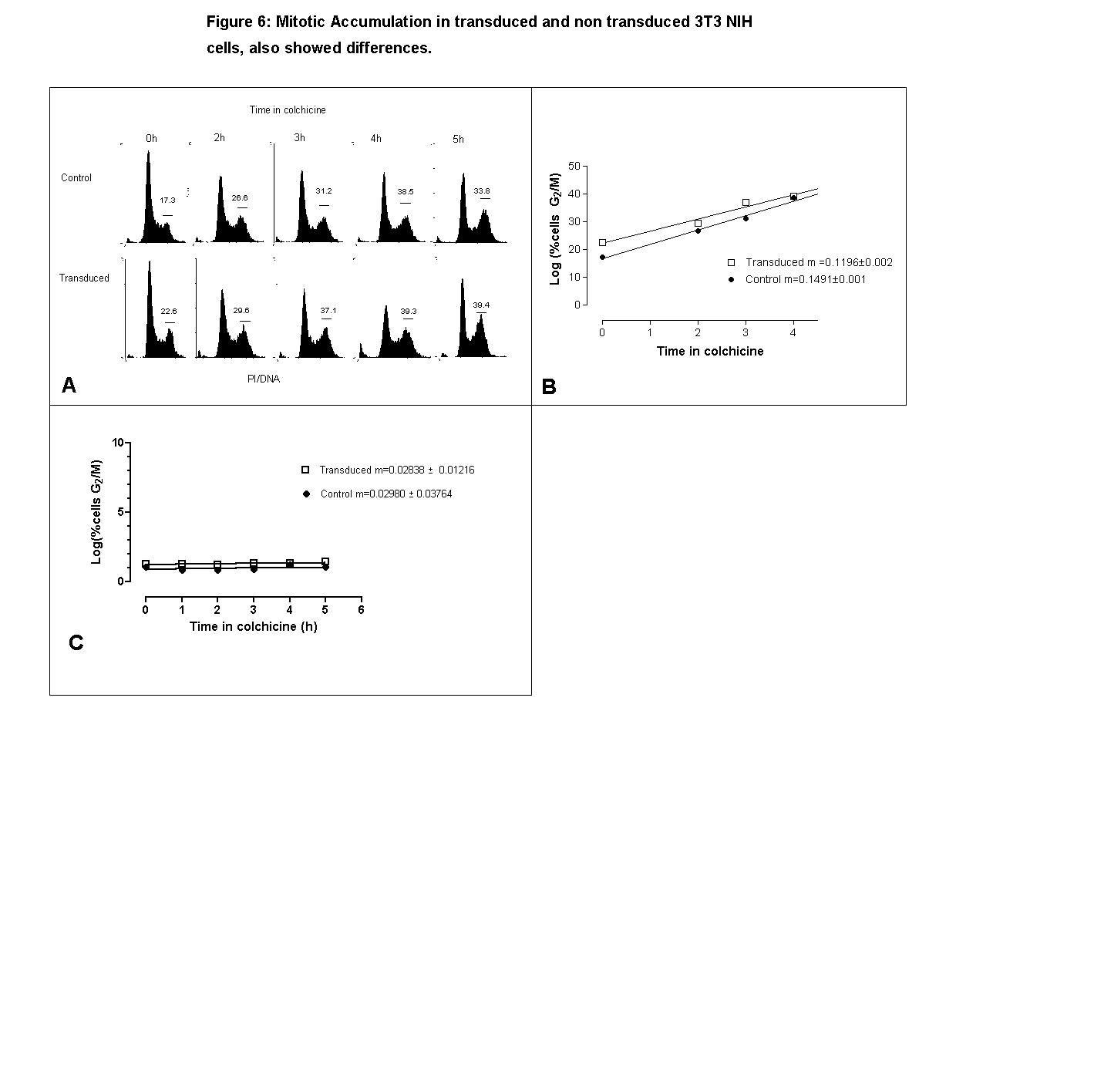{kind=link}
As shown in Fig. 6C, the slopes of the function of mitotic accumulation on 3T3 cells transduced by the PG13-GFP vector and control cells are not statiscally different.
Discussion
Primary human keratinocytes have been successfully modified through chemical, physical and biological methods [21, 22, 23] being retroviral vectors the most utilized. The spectrum and potential of infection of the retroviral vector FOCH-29 NeoR utilized in this study has been demonstrated in a wide range of target cells of human and murine origin [18]. Under the conditions of culture described herein, titles between 4x106 and 3x107 cfu/mL were obtained with which a transduction efficiency of 46.66 percent was reached. Although not optimal, now that we have not reached a 100 percent efficiency in all the experiments, differences on cell cycle were established between transduced and non transduced keratinocyte cultures. Studies performed by other research groups have shown a transduction efficiency between 70 and 100 percent when cultures of human keratinocytes are transduced with vector titles similar to our vector [8, 23, 24, 25, 26] whereas other groups have reached transduction efficiencies similar to ours [23, 27]. Differences on the transduction protocols could explain our results. The experiments of transduction in vitro of human keratinocytes performed in this study took place when cells were between 40 and 60 percent confluent, approximately 72-120 hours post culture. Transduction was unsuccessful in 36 percent of the experiments. A possible explanation was that the time to get the confluence established for transduction was long in these experiments. Cultures were able to become older and mitotically inactive. They became difficult to be modified by our retrovirus, not the result of the vector title or supernatant toxicity. Human keratinocytes were transduced with only one dose of infection, which in practical terms reduces the time of culture with the viral supernatant and diminishes the possibilities of toxic effects of factors present in the infecting supernatant.
The reports of the deleterious effects of genetic modification in the proliferative capacity, the cell cycle and the differentiation of keratinocytes are very few. It is reported that methods of genetic modification such as the liposomes affect human keratinocytes [13]. This has been proven by functional tests like the incorporation of brome deoxiuridine (BrdU) and the percentage of CFE and the expression of proteins like involucrine. Results with retroviral vectors prove that these vectors did not affect them [13]. Because the FOH29-NeoR vector was used for the first time in the modification of keratinocytes in this study, we have attempted to approach this aspect by performing CFE tests. Analysis of the cellular cycle by incorporation of PI and blockade with colchicine was also conducted. In CFE tests, no variation was found in the capacity to form colonies between transduced and not transduced keratinocytes. In our study, it was observed that the greater percentage of colonies present in modified and nonmodified cultures morphologically correspond to meroclones and paraclones colonies type and the presence of holoclones was low or absent (data not shown). This can be attributed to the fact that these cultures correspond to tertiary cultures where keratinocyte stem cells are difficult to find. It is reported that as the culture gets older, a clonal conversion takes place from holoclone to meroclone and from meroclone to paroclone [19]. In the same way, the low yield in CFE in our cultures could be explained by this factor, rather than by the direct effect of genetic modification by the FOCH29NeoR vector; the negative controls of the four experiments had a similar behavior in both the type of colonies and in the percentage of CFE compared to the cultures transduced by one and two doses of infection (Fig. 2).
When the histograms obtained with transduced and nontransduced keratinocytes were compared by using the Kruskal-Wallis analysis, no significant differences were observed in terms of the distribution of cells in the different phases of the cellular cycle. It was observed that there was a time dependent increase in the number of mitotic cells, in the tests of blockade with colchicine in transduced and nontransduced cells. At the beginning of the blockade, modified and nonmodified keratinocytes showed comparable mitotic indexes. The slope of the function of mitotic accumulation obtained by PI incorporation in the transduced cells was significantly smaller (0.36±0.052) than of the non-transduced cells (1.48±0.15). The same results were obtained by Giemsa staining, where the slope of MI in the transduced cells was significantly smaller (0.006±0.0007) than of the nontransduced cells (0.008±0.001). This suggests that the transduced keratinocytes have a delay in the entrance to G2. As far as we know, this is the first report of this effect on modified keratinocyte cell-cycle time attributed to transduction with retroviral-vector induction. One possibile explanation is that vector integration causes DNA damage activating the G2 checkpoint control pathway preventing cell cycle progression. A recent report authors proposes that the integrase-mediated joining of retroviral and host DNA is sensed as a damage by the host cell, and that DNA repair proteins, such as DNA-PK may be required to facilitate stable integration [29]. Other proteins (ATM and ATR kinases) seem to have broader roles than DNA-PK in response to DNA damage, which include the regulation on cell cycle checkpoints. Cell-cycle activation checkpoints are not specific for viral infection. Other factors can also activate them, including ionizing radiation, genotoxics, and mutational agents. Also, it has been demostrated that cell cycle checkpoints response to DNA-damaging agents may vary according to cell type within any given tissue. Exposure to gamma radiation or adriamicine induces fibroblast arrest in G1 whereas the same stimulis induces G2 arrest in keratinocytes [29].
These results bring up many questions with respect to the applications that the observed effect can have on the transduced cells, especially on the keratinocytes. Future studies of synchronization in G2 of modified and control cells and measurement of the time to get off this phase might demonstrate whether the observed effect is correct. If so, transduced cells should take a long time to get off this phase. It would be very interesting to study levels of some proteins known as check-point controls in special proteins involved in G2/M transition.
Appareantly, the delay in the cellular cycle observed in the modified human keratinocytes is not observed by in vitro CFE, and possibly not significant in the total generation time of these cells, but this remains to be confirmed with other retroviral vectors.
The analysis of cellular cycle performed on the 3T3 NIH murine fibroblasts as control showed a clear difference in the proportion of cells in the different phases of the cellular cycle. As with primary keratinocyte cultures, this indicates that the transduced cells have a delay in the entrance to G2 in comparison to the nontransduced cells. This may be the result of an effect of the genetic modification. Although this cell line has been utilized as model in many gene transfer studies, as far as we know this is the first time reports on the cell cycle changes after retroviral modification are done. Control experiments in which another reporter gene was used did not show changes of the cell cycle as a consequence of transduction (Fig. 5B).
Reports on the effect of encoded transgene include the altered morphology and reduced proliferative potencial in G418 selected canine keratinocytes [30]. On the other hand it has been reported that GFP inhibit the growth of retroviral producer cells [31]. However, control experiments on 3T3 cells with the GFP reporter gene could mean that changes found by us on keratinocytes and 3T3 cells are due to the effect of the NeoR gene. Nevertheless, nonselected cells were used on CFE or cell-cycle experiments in our study. This suggests that the observed effect is not a result of the selection with the drug, but rather of the transduction with the FOCH29 vector. The gold-standard control would be the construction and use of FOCH29 vectors with reporter genes not dependent on drug selection.
Because of these findings, future efforts will be aimed at distinguishing between the effects of retroviral transduction, the encoded transgene, and the retroviral backbone on human-keratinocyte retroviral modification. Long-term goals will be the development of experimental protocols and therapeutic alternatives in the treatment of diseases that show a high prevalence in our population, such as skin tumors, extensive burns, and leg ulcers. These patients could be treated with modified skin substitutes that could potentiate the efficiency of the grafts, increasing their functional ability in tissue repair as been shown in several experimental models [32, 33]. If the entrance to G2 is delayed in these skin substitutes through the genetic modification of the keratinocytes, it is reasonable to think that the time to obtain transplantable cultures would be longer, increasing the probability of the cells aging in vitro, thus compromising their potential long-term use. Hence, once grafted, the genetically modified cells would be at proliferative disadvantage with respect to normal keratinocytes rendering the expected beneficial effect of the grafted tissue to be quickly lost. The results of this study allow us to suggest that for each type of vector it would be necessary to determine its effects on the target cell. This type of study can contribute to the elucidation of consequences of gene transfer in order to diminish the risks in clinical application of this technology, and avoid the recently reported deleterious effects in patients in which retroviral vector-modified cells were applied [34].
Acknowledgements: This study was supported by grants from the CODI (Comité para el Desarrollo de la Investigación) University of Antioquia and the Sociedad Colombiana de Dermatología. The authors gratefully acknowledge Dr Alvaro Meana for supplying the 3T3 Swiss cell line, Dr Jose Regueiro for his help with the PG13-GFP producer line, Carolina Ruiz and Carolina Henao for their help in English preparation of the paper and Dr. Luis Fernando García for his critical reading of the manuscript.
References
1. Krueger G, Morgan J, Jorgensen M. Genetically Modified Skin to Treat Disease: Potential and Limitations. J Invest Dermatol 1994: 103:76s-84s. 45.2. Fenjves E. Approaches to Gene Transfer in Keratinocytes . J Invest Dermatol 1994: 103:70s-75s
3. Vailly J, Gagnoux P, Ambra E, et al. Corrective gene transfer of keratinocytes from patients with junctional epidermolysis bullosa restores assembly of hemidesmosomes in reconstructed epithelia. Gene Ther 1998: 5:1322-1332
4. Choate K, Medalie D, Morgan J. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat Med 1996: 2:1263-1267
5. Spirito F, Meneguzzi G, Danos O, Mezzina M. Cutaneous gene transfer and therapy: the present and the future. J of Gene Med 2001: 3:21-31
6. Pauline M, Schwartz R, Blaese M. Keratinocyte gene therapy for adenosine deaminase deficiency: A model approach for inherited metabolic disorders. Hum Gene Ther 1997: 911-917
7. Larcher F, Del Rio M, Serrano F, et al. A cutaneous gene therapy approach to human leptin deficiencies: correction of the murine ob/ob phenotype using leptin-targeted keratinocytes grafts. Faseb J. 2001:15:1529-1538
8. Ghazizadeh S, Carroll JM, Taichman LB. Repression of retrovirus-mediated transgene expression by interferons: implications for gene therapy. J of Virol 1997:71: 9163-9169
9. Ghazizadeh S, Kalish R, Taichman L. Immune-Mediated Loss of Transgene Expression in Skin: Implications for Cutaneos Gene Therapy. Mol Ther. 2003: 7:296-303.
10. Stripecke R, Villares C, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy). Gene Ther. 1999:6:1305-1312.
11. Fenjves E, Shou Nan Y, Kurachi K, Taichman LB. Loss of expresión of a retrovirus transduced gene in Human keratinocytes. J Invest. Dermatol. 1996:106:576-578
12. Ghazizadeh S. Harrington R, Taichman LB. in vivo transduction of mouse epidermis with recombinant retroviral vectors: implication for cutaneous gene therapy. Gene Ther. 1999:6:7:1267-1275
13. Jensen UB, Petersen MS, Lund TB, Jensen TG, Bolund L. Transgene expression in human epidermal keratinocytes: cell cycle arrest of productively transfected cells. Exp Dermatol 2000: 4: 298-310.
14. Dando J, Aiuti A, Deola S, Ficara F, Bordignon C. Optimisation of Retroviral supernatant production conditions for the genetic modification of human CD34+ cells. J Gene Med 2001: 3:219-22
15. Cohen-Haguenauer O, Restrepo LM, M Masset, N Pellerain, JM Heard, M Marty, M Boiron. Construction of a retroviral vector derived from Friend Murine leukemia virus with high infectious titers and evaluation of human CD34+ haematopoietic progenitors transduction potential. Gene Delivery systems, París. OECD, 1996:127-137
16. Check Erika. Regulators split on gene therapy as patient shows signs of cancer. Nature 2002:419:545-546
17. Castro AP, Fernadez M JM, Millan R, Sanal O, Allende L, Regueiro JR. Toward gene therapy of human CD3 deficiencies. Hum Gene Ther. 2003:14:1653-1661
18. Cohen-Haguenauer, O; Restrepo, LM; Masset, M; Bayer, J; Pellerain, N; dal Cortivo, L; Marolleau, JP; Boiron, M; Marty, M. Efficient transduction of haemopoietic CD34+ progenitors of human origin using an original retroviral vector derived from Fr-MuLV: in vitro assessment. Hum Gene Ther. 1998:9:207-216
19. Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci. USA 1987: 84:2302-2306
20. Puck Theodore,J. and Ateffen Jan. Life cycle analysis of mammalian cells. A method for localizing metabolic events within the life cycle, and its application to the action of colcemid and sublethal doses of X-irradiation.. Biophisical Journal 1963:3:379-396.
21. Jiang C, Connolly D, Blumenberg. Comparison of methods for transfection of human epidermal keratinocytes. J Invest Dermatol 1991:97:969-973
22. Kuroki T, Kashiwagi M, Ishino K, Hug N, Ohba M. Adenovirus mediated Gene transfer to keratinocytes. A review. J Invest Dermatol 1999:4:153-157
23. Garlick J, Katz A, Fenjves, Taichman L. Retrovirus-Mediated Transduction of Cultured Epidermal Keratinocytes. J Invest Dermatol 1991:97:824-829
24. Jensen T, Sullivan D, Morgan R, et al. Retrovirus mediated gene transfer of ornithine-delta-aminotransferasa into keratinocytes from gyrate atrophy patients. Hum Gene Ther 1997:8:2125-213215
25. Robbins PB, Lin Q, Goodnough JB et al. in vivo restoration of laminin 5 §3 expression and function in junctional epidermolysis bullosa. Proc Natl Acad Sci. 2001: vol 98. 5193-5198.
26. Kolodka TM, Garlik JA, Taichman LB. Evidence for keratinocyte stem cells in vitro: Long term engraftment and persistence of transgene expression from retrovirus transduced keratinocytes. Proc Natl Acad. Sci. 1998: vol 95: 4356-4361
27. Krueger GG, Jogersen CM, Matsunami N, Morgan JR, et al. Persisten transgene expresión and normal differentiation of immortalized human keratinocytes in vivo. J Invest Dermatol. 1999:112:233-239.
28. Daniel R, Kao G, Taganov K, Greger J, Favorova O et al. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc Natl Acad Sci. 2003:100:4778-4783.
29. Flatt PM, Price JO, Shaw A, Pietempol JA. Differential cell cycle checkpoint response in normal human keratinocytes and fibroblast. Cell Growth Differ. 1998: 9[7]: 535-543
30. Stockschlaeder MA, Storb R, Osborn WR, Miller AD. L-Histidinol provides effective selection of retrovirus-vector-tranduced keratinocytes without impairing their proliferative potential. Hum Gene Ther. 1991:2[1]:33-39.
31. Hanazono Y, Yu JM, Dunbar CE, Emmons RV. Green fluorescent protein retroviral vectors: low titer and high recombination frecuency suggest a selective disadvantage. Hum gene Ther. 1997:8[11]:1313-1319
32. Eming S, Yarmush M, Morgan J. Enhanced Function of cultured epithelium by genetic modification: cell-based synthesis and delivery of growth factors. Biotech and Bioeng 1996:52:15-23.
33. Del Rio M, Larcher F, Meana A, Segovia J, Alvarez A, Jorcano J. Nonviral transfer of genes to pig primary keratinocytes. Induction of angiogenesis by composite grafts of modified keratinocytes overexpressing VEGF driven by a keratin promoter. Gene Ther 1999:6:1734-1741
34. Hecein-Bey-Abina S, von Kalle C, Schmidt M, et al. A serius adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl. J. Med. 2003:348:255-256.
© 2005 Dermatology Online Journal