CD4+/CD56+ Hematodermic neoplasm (plasmacytoid dendritic cell tumor)
Published Web Location
https://doi.org/10.5070/D37n62p0fjMain Content
CD4+/CD56+ Hematodermic neoplasm (plasmacytoid dendritic cell tumor)
Michael Shiman, Robb Marchione MD, Carlos Ricotti MD, Paolo Romanelli MD, Javier Alonso-Llamazares MD
Dermatology Online Journal 14 (11): 5
Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, Florida. Jalonso2@med.miami.eduAbstract
A 60-year-old male presented with multiple, purplish-red, nodules and plaques. After a complete work-up, he was diagnosed with CD4+/CD56+ hematodermic neoplasm. We review the clinical, pathological, and immunohistochemical features of this disease. CD4+/CD56+ hematodermic neoplasm, which is also known as blastic natural killer-cell lymphoma, is a rare, aggressive neoplasm with a strong predilection for skin involvement.
Clinical synopsis
A 60-year-old Hispanic male was admitted for evaluation of multiple cutaneous lesions. The patient had been in his usual state of health until five months prior when he developed pruritus around his right knee. Over the next several days, he developed multiple, purplish-red, nodules and coalescent plaques on his lower extremities, trunk, and face. He denied any constitutional symptoms.
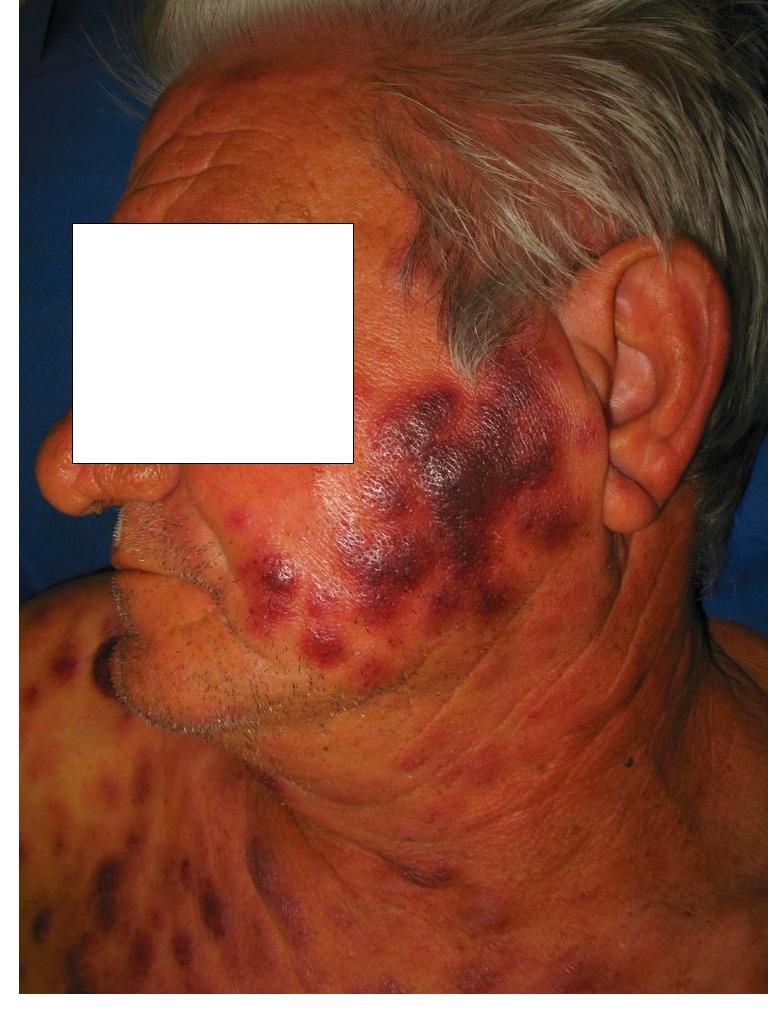 |
| Figure 1 |
|---|
| Figure 1. Multiple, purplish-red, indurated nodules on the face |
Physical exam on admission revealed multiple, well-demarcated, contusiform, indurated nodules and plaques scattered over his legs, arms, trunk, and face. Axillary lymphadenopathy was present. Laboratory results revealed hemoglobin 12.7 g/dL, white blood cell count 6.0 x 109/L with 55 percent lymphocytes, and platelet count of 241 x 109/L. Lactate dehydrogenase was elevated at 1139 U/L. Chest radiography was unremarkable and computed tomography of the chest and abdomen revealed axillary, mesenteric, retroperitoneal, pelvic, and inguinal lymphadenopathy.
Skin and axillary biopsies were obtained and revealed monomorphous, non-epidermotrophic, medium-to-large round cells with pleomorphic nuclei. The infiltrate was confined to the dermis and separated by the epidermis with a Grenz zone.
Immunohistochemistry identified CD4+ and CD56+ cells. Cells were negative for the following: CD3, CD8, CD20, CD79, myeloperoxidase, and Epstein-Barr virus antigen. PCR assays revealed no clonal B or T cell populations. A bone marrow biopsy showed mildly hypercellular marrow consisting of an infiltrate resembling CD56+ blast cells. Based on these findings, the patient was diagnosed with CD4+/CD56+ hematodermic neoplasm.
The patient initially received a cycle of hyperfractionated cyclophosphamide, hydroxydaunomycin, vincristine, plus etoposide with little response. The patient then received four cycles of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (hyper-CVAD) fortified with methotrexate and cytarabine. Following the second cycle of hyper-CVAD the lesions achieved near complete resolution. Clinical exam after the second cycle revealed diffuse hyperpigmented macules and patches with pathology showing post-inflammatory pigment alteration.
 | 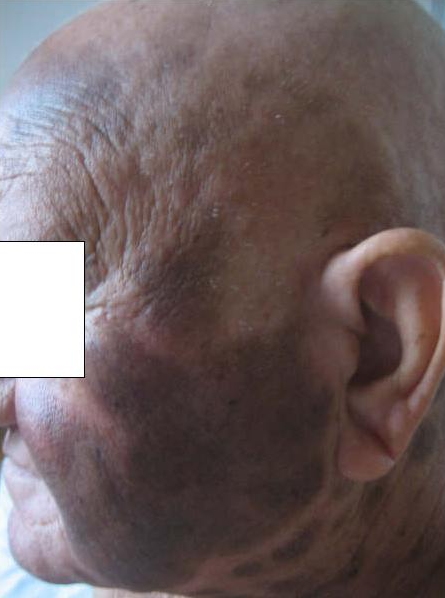 |
| Figure 2 | Figure 3 |
|---|---|
| Figure 2. Biopsy specimen revealing a dense, diffuse infiltrate throughout the dermis, with a grenz zone sparing the dermal-epidermal
junction. Figure 3. Clinical findings post treatment, revealing post-inflammatory hyperpigmented lesions. | |
Discussion
CD4+/CD56+ hematodermic neoplasm, also known as blastic natural killer-cell lymphoma, is a rare, distinct malignancy with a strong predilection for cutaneous involvement [1]. Once believed to originate from a natural killer cell precursor, more recent immunohistochemical marker analysis suggests a plasmacytoid dendritic cell lineage [2].
This case illustrates the importance of recognizing the clinical, pathological, and immunohistochemical features of this disease so that the appropriate treatment can be considered. This neoplasm is rare, aggressive, and has a median survival of 12 to 14 months [3]. Expression of the cell surface marker CD123 (IL-3 Receptor α) is useful in helping to reach the diagnosis [4]. In fact, it may help to separate this entity from the cutaneous involvement of myelomonocytic leukemia which may precede the leukemic phase of the disease [5]. An initial promising response to chemotherapy is common, but relapses and drug resistance frequently occur [3]. The origin of this neoplasm is now believed to be plasmacytoid dendritic cells. These are hematopoietic-derived cells that produce regulatory cytokines and regulate the innate and adaptive cell immunity [4]. A recent case report demonstrated rapid clinical response to pralatrexate, a new investigational antifolate [3]. Further studies and trials are needed in order to find a promising treatment option for this rare, aggressive neoplasm.
References
1. Martin JM, Nicolau MJ, Galan A, Ferrandez-Izquierdo A, Ferrer AM, Jorda E. CD4+/CD56+ haematodermic neoplasm: A precursor haematological neoplasm that frequently first presents in the skin. J Eur Acad Dermatol Venereol. 2006;20(9):1129-1132. PubMed2. Feuillard J, Jacob MC, Valensi F, et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002;99(5):1556-1563. PubMed
3. Leitenberger JJ, Berthelot CN, Polder KD, et al. CD4+ CD56+ hematodermic/plasmacytoid dendritic cell tumor with response to pralatrexate. J Am Acad Dermatol. 2008;58(3):480-484. PubMed
4. Pina-Oviedo S, Herrera-Medina H, Coronado H, Del Valle L, Ortiz-Hidalgo C. CD4+/CD56+ hematodermic neoplasm: Presentation of 2 cases and review of the concept of an uncommon tumor originated in plasmacytoid dendritic cells expressing CD123 (IL-3 receptor alpha). Appl Immunohistochem Mol Morphol. 2007;15(4):481-486. PubMed
5. Petrella T, Bagot M, Willemze R, et al. Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic neoplasms). A review. Am J Clin Pathol 2005;123:662-675. PubMed
© 2008 Dermatology Online Journal