Hepatitis C Virus infection, type III cryoglobulinemia, and necrotizing vasculitis
Published Web Location
https://doi.org/10.5070/D36m82n0j1Main Content
Hepatitis C Virus infection, type III cryoglobulinemia, and necrotizing vasculitis
Isaac Brownell MD PhD, William Fangman MD
Dermatology Online Journal 13 (1): 6
New York University Department of DermatologyAbstract
A 53-year-old man with chronic hepatitis-C virus infection presented with livedo reticularis, purpura, and leg ulcers. A skin biopsy specimen showed a necrotizing vasculitis. The skin biopsy specimen and serology confirmed the diagnosis of type-III cryoglobulinemia. Bone marrow and peripheral blood showed proliferation of atypical CD5-positive B cells that included a monoclonal population. There is growing evidence that chronic hepatitis-C infection can result in immune dysregulation and expansion of autoimmune B cells that produce cryoglobulins.
Clinical synopsis
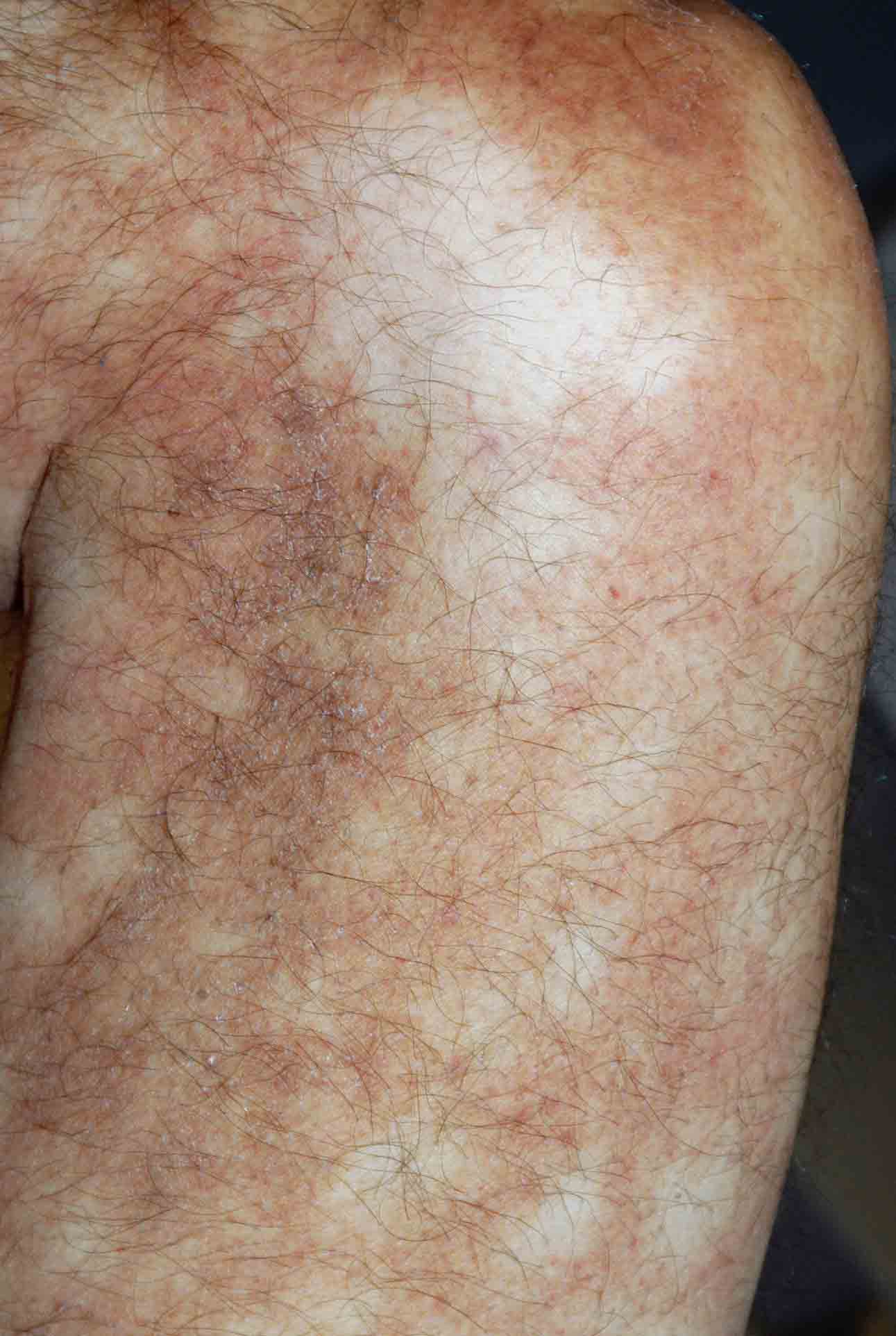
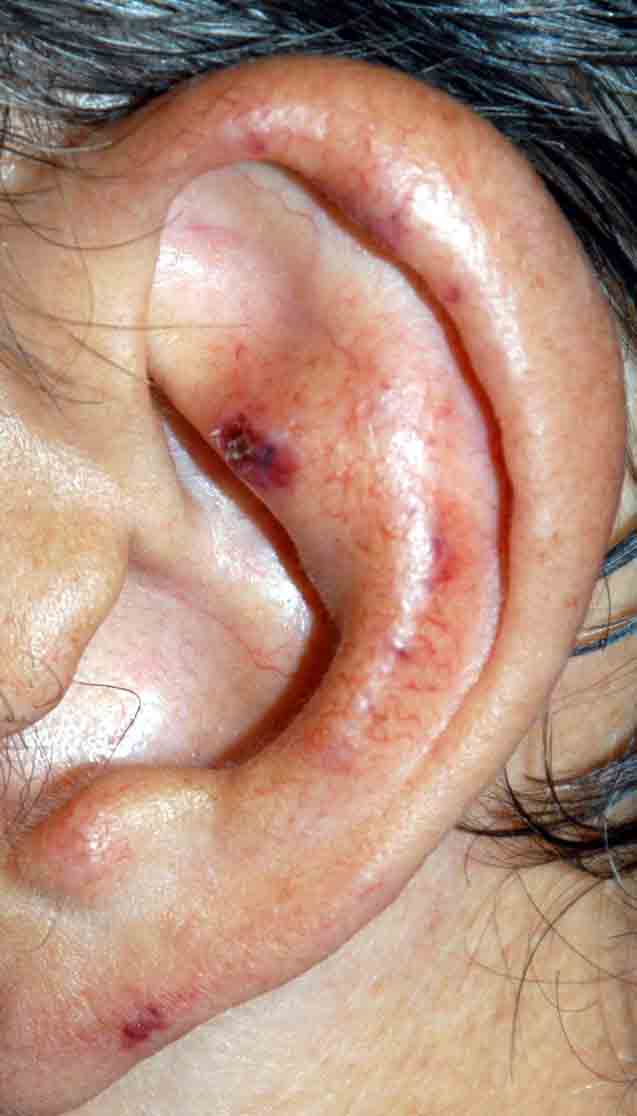
A 53-year-old man presented with multiple, painful ulcers arising spontaneously within an erythematous, livedoid eruption on the distal aspects of his legs. The eruption spread to involve the upper arms, and the patient developed purpura on his limbs, ears, and abdomen. The onset occurred during a winter month in New York City. General fatigue and a symmetric loss of sensation in the lateral portions of the feet accompanied the condition. The patient denied fevers, arthritis, Raynaud phenomenon, abdominal pain, and visual disturbances.
Over 6 months, the ulcers have improved with wound care and the intermittent use of topical gentamicin and oral ciprofloxacin to control bacterial colonization and tissue infection. No new ulcers have formed. The livedoid eruption and purpura fluctuate in severity while the fatigue and neuropathy remain stable.
Medical history includes chronic hepatitis C (genotype 3a) and a past intravenous drug abuse. The only medication was methadone. There is no personal or family history of renal failure, coagulopathy, or autoimmune disease.
Reticulated, erythematous, hyperpigmented patches with areas of petechiae were present over the distal portions of the thighs and upper arms. Multiple, 1-9 cm, oval or angulated ulcers with eschars, surrounding erythema, and hyperpigmentation were noted on the ventral and dorsal portions of the legs below the knees and on the dorsal aspects of the feet. Purpuric macules and papules were scattered symmetrically over the forearms, abdomen, and ears. Fingers and toes were clear. Peripheral pulses were intact and symmetric bilaterally. There was no palpable lymphadenopathy or hepatomegaly. Blood pressure was normal.
 |  |
| Figure 1 | Figure 2 |
|---|
A complete blood count demonstrated a lymphocytosis with 12.1 K/mm³ and 51 percent atypical lymphocytes. A comprehensive metabolic panel, hepatic function panel, and 24-hour urine protein were normal. Rheumatoid factor was elevated at 171 IU/ml. Cryoglobulins were present, and immunoglobulin typing by serum protein electrophoresis and immunofixation of the cryoprecipitate showed polyclonal IgM against polyclonal IgG (type-III cryoglobulins). Serum IgG was normal at 955 mg/dl, while IgM was elevated at 359 mg/dl. Erythrocyte sedimentation rate was elevated at 28 mm/hour. Hepatitis-C RNA was present at 8,800 copies/ml. LDH was elevated at 198 U/L. Bone marrow biopsy and serum flow cytometry showed a CD5 positive, CD19 positive, cyclin D1 negative proliferation of atypical lymphocytes. A circulating population of HLA DR monoclonal κ-positive B cells was identified. Cell morphology and immunophenotype analysis was most consistent with a chronic lymphoproliferative autoimmunity related to hepatitis-C infection rather than malignancy. Antinuclear antibody, C-antineutrophil cytoplasmic antibodies (ANCA), P-ANCA, anti-DNA antibody, anti-phospholipid antibody, α-fetoprotein, and human immunodeficiency virus tests were negative. Computed tomography scans showed splenomegaly at 14 cm and reactive retroperitoneal lymphadenopathy.
 |
| Figure 3 |
|---|
On histopathology, within the dermal and subcutaneous vascular lumens are thrombi consisting of an amorphous pink material. The thrombi stain bright red with periodic acid-Schiff stain. Immunohistochemistry study shows that the thrombi are positive for both kappa and lambda antibodies. In addition, they are positive for IgG and IgM.
Comment
Cryoglobulins are protein complexes that reversibly precipitate when cooled below body temperature. The immunoglobulins found in the cryoprecipitate define the type of cryoglobulinemia [1]. Type-I cryoglobulins are monoclonal antibodies that complex with serum proteins. They are most commonly monoclonal IgM but can also be IgG or rarely IgA or immunoglobulin light chains. The mixed cryoglobulins, types II and III, are rheumatoid factors—IgM antibodies that recognize autologous-polyclonal IgG. Type-II cryoglobulins are monoclonal IgM rheumatoid factors; type III cryoglobulins are polyclonal [1, 2, 3].
Type I cryoglobulinemia is almost exclusively associated with lymphoproliferative disorders, such as multiple myeloma, chronic lymphocytic leukemia, and Waldenstroem macroglobulinemia. Mixed cryoglobulinemia (MC) is frequently associated with chronic infections, autoimmune disease, or hematologic malignant conditions. When an etiologic entity is not identified, the term essential-mixed cryoglobulinemia is used [2, 3].
Clinically MC is a small vessel, immune-complex mediated vasculitis. Skin manifestations include palpable purpura, livedo reticularis, Raynaud phenomenon, leg ulcers, acrocyanosis, and cold urticaria. Extracutaneous symptoms include arthralgia, asthenia, peripheral neuropathy, and nephropathy. Although purpura, asthenia, arthralgia, and sensory neuropathy are the most common findings in symptomatic patients, the majority of patients with circulating cyroglobulins show no clinical disease [2, 3]. The differential diagnosis of MC comprises other occlusive vasculopathies, such as hypercoagulable conditions, septic vasculitis, disseminated intravascular coagulopathy, thrombotic thrombocytopenic purpura, coumadin skin necrosis, and calciphylaxis.
In recent years it has become clear that in some populations the majority of mixed cryoglobulinemia is found in patients infected with the hepatitis C virus (HCV). Rates of HCV infection in MC range from 30 to 96 percent, depending on the geographic location. Conversely, cryoglobulins were found in 29-54 percent of patients with HCV, with type-II MC more common than type III. Furthermore, anti-HCV antibodies and HCV RNA are frequently found in MC cryoprecipitate at concentrations higher than they are in the serum. The theory that there is a causal association between HCV and MC is supported by the observation that treatment of HCV often improves MC [2, 3, 4].
It is believed that chronic HCV infection causes an immune dysregulation that results in a polyclonal expansion of CD5 positive B-cells in the liver, blood, or bone marrow. These cells are thought to produce the polyclonal IgM rheumatoid factors found in type III MC. The emergence of a clonal B-cell population could then lead to clonal heterogeneity and ultimately a monoclonal production of IgM as seen in type-II MC [3, 4].
In our patient, a clonal B-cell population was detected in the blood; however, protein electrophoresis failed to show an IgM monoclonal κ band. Similar HCV patients with type-III MC and clonal B-cell proliferations are not uncommon [4]; however, it remains unclear if this actually represents a transition state between type-III and type-II MC. Although no lymphoproliferative malignant condition was detected in our patient, HCV patients with symptomatic MC may be more likely to eventually develop a low-grade lymphoma or B-cell malignant condition [3, 4].
Treatment of MC is dependent on the severity of disease and etiologic factors. Any underlying infection, malignant condition, hematologic disorder, or autoimmune condition should be treated appropriately. Arthralgias often respond to nonsteroidal anti-inflammatory drugs. Low-dose glucocorticoids can be used for minor or intermittent symptoms in the skin, joints, and sensory nervous system. Rapidly progressive vasculitis or glomerulonephritis requires high-dose glucocorticoids, cytotoxic agents, and plasmapheresis. The association of MC and HCV has made interferon plus ribavirin a mainstay of treatment for MC. Clearance of virus correlates well with reductions of CD5-positive B-cell proliferations and circulating cryoglobulins. Unfortunately relapse rates are high after discontinuation of therapy [2, 3]. The use of the anti-CD20 drug rituximab to target B-cells has recently shown efficacy in controlling MC in selected patients [5]. Our patient plans to begin a regimen of pegylated interferon α 2a and ribavirin to treat his hepatitis C and its associated lymphoproliferative condition with cryoglobulinemia.
References
1. Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M.. Biologic and clinical significance of cryoglobulins: a report of 86 cases. Am J Med 1974;57:775. PubMed2. Cacoub P, Costedoat-Chalumeau N, Lidove O, Alric L. Cryoglobulinemia vasculitis. Curr Opin Rheumatol. 2002 Jan;14(1):29-35. PubMed
3. Sansonno D, Dammacco F. Hepatitis C virus, cryoglobulinaemia, and vasculitis: immune complex relations. Lancet Infect Dis 2005;5:227. PubMed
4. Vallat L, Benhamou Y, Gutierrez M, Ghillani P, Hercher C, Thibault V, Charlotte F, Piette JC, Poynard T, Merle-Beral H, Davi F, Cacoub P. Clonal B cell populations in the blood and liver of patients with chronic hepatitis C virus infection. Arthritis Rheum. 2004 Nov;50(11):3668-78. PubMed
5. Zaja F, De Vita S, Mazzaro C, Sacco S, Damiani D, De Marchi G, Michelutti A, Baccarani M, Fanin R, Ferraccioli G. Efficacy and safety of rituximab in type II mixed cryoglobulinemia. Blood. 2003 May 15;101(10):3827-34. Epub 2003 Jan 30. PubMed
© 2007 Dermatology Online Journal