Immunohistochemistry screening of sebaceous lesions for Muir-Torre syndrome in a 26-year period in a Mexican population
Published Web Location
https://doi.org/10.5070/D3666004s1Main Content
Immunohistochemistry screening of sebaceous lesions for Muir-Torre syndrome in a 26-year period in a Mexican population
Carla Archer-Dubon1, Bertha Alvarez-Zavala1, Edgardo Reyes2, Rocio Orozco-Topete1
Dermatology Online Journal 14 (12): 1
1. Dermatology Department, Instituto Nacional de Ciencias Médicas y Nutrición, Mexico City, Mexico. carla_archer@yahoo.com2. Pathology Department, Instituto Nacional de Ciencias Médicas y Nutrición, Mexico City, Mexico
Abstract
Muir-Torre syndrome (MTS) is an autosomal dominant genodermatosis defined as the association of rare sebaceous gland skin tumors, keratoacanthomas, and a personal or familial history of malignant visceral tumors. Germline mutations in certain mismatch repair genes (MMR) have been identified in MTS families and their identification is a cornerstone for diagnosis of MTS. We reviewed our series of sebaceous neoplasms and performed immunohistochemistry (IHC) in order to screen for new MTS cases. Sebaceous neoplasms and visceral tumors from the same patient diagnosed between 1980-2006 were included. Immunohistochemistry to determine the presence or absence of MMR gene products in skin and visceral tumors was performed with mouse monoclonal antibodies anti-MSH2, anti-MSH6 and anti-MLH1. Six sebaceous neoplasms were identified in six females. Four patients presented a lack of expression of at least one of the MMR proteins in visceral and cutaneous neoplasms, thus warranting the diagnosis of MTS. Immunohistochemistry is a useful and accessible technique for the characterization of MMR gene expression in patients with sebaceous neoplasms.
Introduction
In 1967-8, Muir et al. [1] and Torre D. [2] independently described the association of sebaceous gland tumors, keratoacanthomas (KA), and visceral malignancy, which thereafter was known as the Muir-Torre syndrome. An autosomal dominant inheritance was later established by Reiffers et al. [3] in 1971, ten years later Lynch and colleagues [4] reported four families with colorectal cancer and sebaceous neoplasms. They suggested that Muir-Torre syndrome was a clinical variant of hereditary non-polyposis colorectal cancer (HNPCC). Schwartz and Torre in 1995 [5] defined the disease as the simultaneous appearance of certain sebaceous neoplasms, with or without keratoacanthomas, with one or more visceral malignancies in the absence of a predisposing factor. Germline mutations in the MSH2 and MLH1 DNA mismatch repair genes were thereafter identified by Kruse et al. [6] and Bapat et al. [7], respectively. Currently a diagnosis of Muir-Torre syndrome (MTS) can be confirmed in a patient with concurrent multiple keratoacanthomas or a sebaceous neoplasm, internal malignancies, and a personal or family history of MTS [5, 8, 9].
Confirming the diagnosis of MTS is relevant because patients and their families are at risk for multiple primary malignancies and dermatologists play an important role in diagnosing associated skin lesions. Solitary or multiple sebaceous adenomas are rare and currently considered the most significant cutaneous clue of MTS [5]. Other sebaceous tumors associated with MTS are sebaceous epithelioma, basal cell carcinoma with sebaceous differentiation, and sebaceous carcinoma (especially with a keratoacanthoma-like pattern) [5]. Skin lesions may precede the appearance of internal malignancy that is usually colonic, although there may be a hiatus of many years before both are present; this results in a delayed diagnosis if the treating physician is unaware of this syndrome.
Muir-Torre syndrome is a phenotypic variant of hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome. Most tumors related to HNPCC show a defective DNA mismatch repair (MMR) system, specifically germline mutations in MLH1, MSH2 [6, 7] and MSH6 [10, 11]. These genes are responsible for the repair of concordance errors during DNA replication and their mutations favor microsatellite instability in cutaneous and visceral tumors [12-24]. Microsatellite instability is evident in more than 95 percent of HNPCC-associated neoplasms. This biomolecular alteration has also been observed in sebaceous lesions and KA from MTS patients, thus substantiating a common pathogenesis of colorectal and cutaneous lesions [14, 20].
Most skin tumors from MTS patients express microsatellite instability [12]. Screening for MMR gene defects can be performed by immunohistochemical (IHC) analysis and genotyping for microsatellite instability. Gene sequencing in search of the mutation is diagnostic. Both IHC and genotyping methods can be used to detect mismatch-repair deficiency and are much less expensive methods than gene sequencing. Immunohistochemical analysis, as the primary screening method, has the advantage that it can be routinely performed in a general pathology laboratory, unlike genotyping that requires a molecular laboratory. Recent studies have shown its efficacy in the identification of MMR gene expression in cutaneous lesions of MTS patients and its utility as a screening procedure in patients with a doubtful diagnosis [13, 14, 21, 22, 23, 24]. On the other hand, gene sequencing is difficult, expensive, time-consuming, and rarely accessible to physicians in developing countries such as our own.
Our institution is a tertiary care center and at the Dermatology Department we currently see approximately 6000 patients/year. We designed the following study to determine the number of sebaceous neoplasms in our population base and to detect the mismatch-repair deficiency associated with MTS in these neoplasms irrespective of personal or familial history of visceral malignancy.
Material and Methods
We reviewed all histopathological skin samples from 1980 to 2006 and included all neoplasms with the exception of sebaceous hyperplasias and nevus sebaceus of Jadassohn. All cutaneous tumors or visceral malignancies from the same patient were included, if present. The medical records of these patients were carefully reviewed for personal and family history of visceral malignancies. All procedures were approved by the Ethical Committee of the hospital.
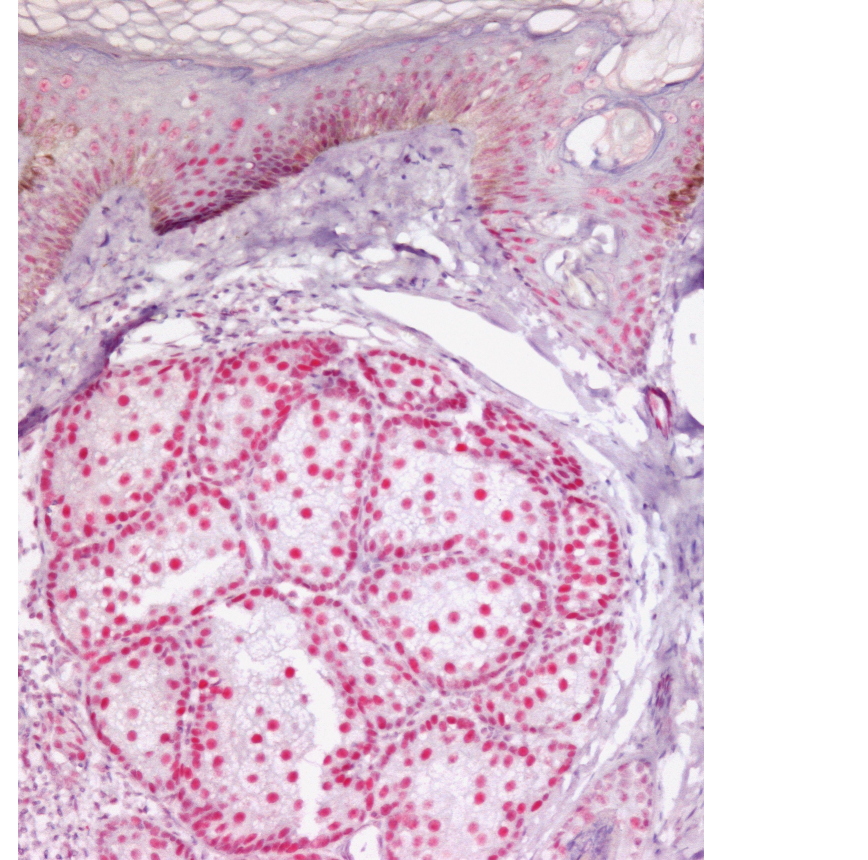 | 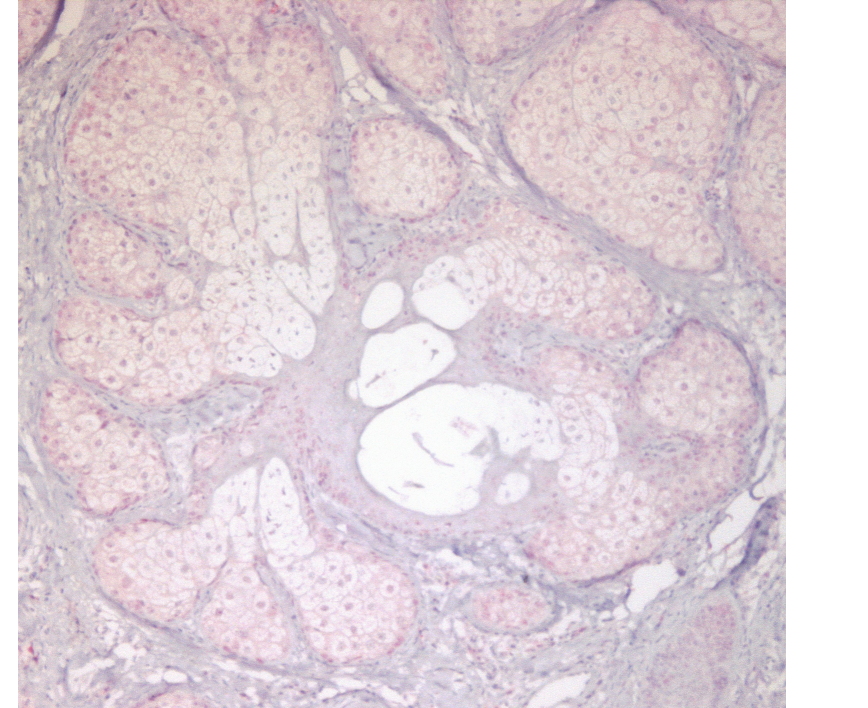 |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. A sporadic sebaceous adenoma with a positive immunohistochemistry for MLH1 (Alcaline Phosphatase red detection kit,
Ventana and conventional H&E stains, x40) Figure 2. A sebaceous adenoma from a Muir-Torre patient with a negative immunohistochemistry for MSH6 (Alcaline Phosphatase red detection kit, Ventana and conventional H&E stains, x40) | |
Standard IHC was performed on tissue from skin and visceral neoplasms to determine the presence or absence of MMR proteins using the following mouse antibodies at the recommended dilutions: anti-hMLH1, anti-hMSH2 and anti-hMSH6 (Pharmingen International, California). Normal gut and skin were included as controls. Stained slides were evaluated by one of us (ER) who was blinded to the clinical data and family history of the patient. After evaluation, slides were unblinded to correlate with the clinical diagnosis and immunohistochemical results.
Results
We found eight sebaceous neoplasms from a total of 13,000 skin biopsies in the period studied; of these, six were available for IHC. A total of thirteen IHC tests were performed on the following lesions: six sebaceous neoplasms (five adenomas, one sebaceous carcinoma), two colon cancers, three cutaneous malignancies (two basal cell carcinomas, one squamous cell carcinoma) and two benign cutaneous tumors (one keratoacanthoma and one intradermal nevus.) (See Table 1) All patients were females aged 38-74 years.
Four of the six sebaceous neoplasms revealed a loss of expression of at least one of the MMR proteins: MLH1 (1), MSH2 (1), MSH6 (2). Of the MMR genes studied, absence of MLH1 was seen in five tumors (sebaceous adenoma -SA- , keratoacanthoma, BCC, colon cancer from patient 1 and sebaceous carcinoma -SC- from patient 3), absence of MSH2 in four tumors (keratoacanthoma, sebaceous adenoma, SCC, and colon cancer) and absence of MSH6 in four tumors (keratoacanthoma, sebaceous adenoma (2) and colon cancer).
Two SA patients presented a coexistence of sebaceous neoplasms and colon cancer in the absence of a predisposing factor (Cases 1 and 2) and first degree relatives with visceral malignancies (colon, stomach, ovary); thus a new diagnosis of MTS was made. In both, the visceral neoplasm preceded the sebaceous neoplasm. These patients were already under oncologic surveillance and their first degree relatives were referred to the Oncology Department. In both, tissue was available from their sebaceous neoplasms, their visceral malignancy and a non-sebaceous cutaneous malignancy (BCC and SCC, respectively). Interestingly, both the non-sebaceous, cutaneous malignancies expressed the same lack of MMR protein (MLH1 and MSH2, respectively) as their sebaceous tumor and visceral malignancy, suggesting a germline mutation for that gene. In contrast, patient 4 had an SA negative for MSH6, but a melanocytic lesion that expressed all three MMR proteins normally. This patient had no family history of cancer.
The SC patient (Case 3) had a first degree relative with colon cancer but no personal history of visceral malignancy to date of study. Both latter patients (cases 3 and 4) are considered probable MTS patients and colonoscopies were performed and were reported to be normal. They will continue with biannual colonoscopies. Patients 5 and 6 had SA with no MMR gene defect detected by IHC. Patient 5 also had a non-sebaceous, cutaneous tumor that expressed all three MMR proteins normally. Remarkably, she has an important familial history of gastrointestinal tract and other visceral malignancies.
Two other SA patients were identified but are not shown because we were unable to perform IHC: one SA in a 33-year-old female with a jejunum cancer and positive first-degree relatives for cancer of duodenum, colon and stomach. Tissue from her SA was unfortunately unavailable. Nonetheless, she meets clinical criteria for Muir-Torre syndrome and she and her available first-degree relatives are under ongoing oncologic surveillance.
A second patient with an SA not included was a 58-year-old male with a renal transplant who subsequently developed gastric cancer and died. First-degree relatives were unavailable. Sebaceous adenomas in the context of immunosuppression are difficult to interpret and more so in cases like this patient who eventually died of a GI tract, albeit not colonic, malignancy.
Discussion
We found eight patients with sebaceous neoplasms, which represents an incidence of 0.06 percent. Six of these were studied by IHC for MMR proteins associated with Muir-Torre syndrome, a variant of Lynch syndrome. An absence of at least one MMR protein was seen in four patients, thus further confirming a diagnosis of MTS. Two of these patients had a lack of the same MMR gene in all of their tumors, suggesting a germline mutation for that particular gene.
MTS is considered an uncommon syndrome. However, based on our results we believe it is much more frequent; we found that 50 percent of the sebaceous neoplasms seen over a 26-year period have immunohistochemical findings consistent with a diagnosis of MTS. MTS is probably being underdiagnosed; how many patients with a "sporadic" colorectal or GI tract malignancy have a concurrent undiagnosed sebaceous neoplasm? Cutaneous clues for the diagnosis of MTS may be subtle and go unrecognized. Although the presence of a sebaceous neoplasm is not sufficient to diagnose MTS, we believe its presence is enough to warrant serious consideration, especially if the sebaceous tumor is uncommon and the patient has a family history of visceral malignancy (particularly of the GI or genito-urinary tract). Any sebaceous neoplasm, excluding sebaceous hyperplasia or nevus sebaceus of Jadassohn, should compel the clinician to rule out MTS, particularly since MTS patients and their immediate families require periodic colonoscopies. MTS patients have at least twice the risk of developing an internal malignancy as compared to the normal population. Other than clinical data and immunohistochemistry, MTS can also be diagnosed by identifying microsatellite instability (MSI) and gene sequencing procedures. However, these are far from being readily accessible to health care providers in developing countries because they are much more expensive and time-consuming; they require specially-equipped laboratories and trained personnel.
Immunohistochemistry has been suggested by several authors as a screening test for MTS since it is a less expensive procedure and available to any general pathology laboratory. In a recent report by Hampel et al, immunohistochemical analysis showed a 93.2 percent sensitivity (95 percent confidence interval) for the detection of abnormalities in at least one protein in colonic tumors of patients with HNPCC. The sensitivity of immunohistochemical analysis to pinpoint the affected gene in tumors with deleterious mutations was high: MSH2 was detected in 11 of 12 tumors, MLH1 in 4 of 5, MSH6 in 3 of 3, and PMS2 in 2 of 2 [24].
Although MSH6 mutations have been reported to be less frequent than MLH1 and MSH2 in Lynch syndrome, MSH6 mutations were identified in 2/6 of our cases. These are associated with certain differing characteristics. They have been related to increased genitourinary cancer and tumors of the endometrium that have been specifically identified in females with lack of expression of MSH6. Studies have reported that female MSH6 mutation carriers have a 73 percent incidence of endometrial carcinoma, compared to 29 percent in MSH2 mutation and 31 percent in MLH1 mutation carriers [25, 26]; Suchy J et al. described a Polish MSH6 family that presented with an endometrial type of ovarian cancer, colon cancer, and endometrial cancers [27]. Thus several authors concur that colorectal cancer cannot be considered obligatory to define Lynch syndrome in these patients. Another feature of MSH6 mutation carriers is the delayed age of cancer onset and incomplete penetrance compared to carriers of MSH2 and MLH1 mutations. Wagner et al. found that colorectal cancer was significantly less frequent in a large Dutch family with atypical HNPCC and an MSH6 mutation when compared to families with mutations in MSH2 or MLH1 (p<0.001) [28]. Berends et al. [11] in 2002 concluded that MSH6 mutation analysis should be considered in all patients suspected to have HNPCC. Neither microsatellite instability nor IHC should be a definitive selection criterion for MSH6 mutation analysis and specific genetic testing is in order for these patients.
Barnetson [29] et al. found a 4.3 percent incidence of MMR gene mutations (15 in MLH1, 16 in MSH2, and 7 in MSH6) among a large series of patients under 55 years of age recently diagnosed with colon cancer, regardless of family history. Carrier frequencies in men (6%) and women (3%) differed significantly (p<0.04). Survival among carriers was not significantly different from that among non-carriers.
One of the sebaceous neoplasms we encountered was found in a renal transplant patient who subsequently developed gastric cancer and died. Others have previously suggested that the clinical manifestations of MTS may be incomplete [5] and immunosuppression may elicit their presentation [30], which could be the case in this patient. As is, the case fulfills clinical criteria for the diagnosis of MTS. Unfortunately we were unable to obtain tissue for immunohistochemistry and his family members were unavailable for further study.
Sebaceous adenomas may present as sporadic lesions not related to MTS, although we believe these lesions are infrequent enough to warrant further investigation with immunohistochemistry for MMR protein expression. Such is the case of one of our patients (Patient 5, Table 1) whose adenoma expressed all MMR proteins normally; thus, theoretically at least, she does not have MTS. Ideally, tissue from her family members with visceral malignancy should be immunohistochemically studied to determine MMR protein expression. We found no consensus regarding follow-up in these cases; given her important family cancer history, we will continue to maintain close oncologic surveillance.
We found eight lesions in a large series of 13,000 skin biopsies in a 26-year period, which is low compared to other series, such as the one reported by Ponti et al. [16] who found 59 lesions (excluding keratoacanthomas) in a 14-year period. This could be explained because we consulted only the records of the Dermatology Department because skin lesions in our setting are mostly excised by dermatologists. Nonetheless, we are aware that occasionally other specialists may biopsy a cutaneous lesion and those would not be accounted for in our series.
Immunohistochemistry is particularly useful in the case of a patient with an unusual sebaceous neoplasm (i.e., not sebaceous hyperplasia) and no personal or familial visceral malignancy. Such is the case of one of our patients (patient 4, Table 1), a 38-year-old female with a sebaceous adenoma that lacked expression of MSH6. Her family history is irrelevant and she has obesity, hypertension, polycystic ovaries, colonic diverticula, internal hemorrhoids, and fibrocystic mastopathy. Upon diagnosing the sebaceous adenoma, a colonoscopy was performed and was reported as normal. Although visceral malignancy has not been diagnosed, she will continue to be under oncologic surveillance.
In conclusion, immunohistochemistry heightened the suspicion of MTS by demonstrating the lack of at least one MMR protein in four of our six patients studied (66%). Genetic counseling and biennial colonoscopies were offered to patients and their families. Also, lifelong surveillance for colorectal cancer, endometrial cancer, and other tumors is indicated.
Our results are, to our knowledge, the first reported in the Mexican population. We concur with other authors in that any sebaceous tumor, excluding sebaceous hyperplasias and nevus sebaceus of Jadassohn, should be studied with immunohistochemistry to determine MMR protein status. Immunohistochemistry is probably available to any general pathologist in a developing country and its sensitivity to detect affected genes has been proven. Nonetheless, a careful, personal and family history of visceral malignancy in addition to a thorough dermatologic evaluation are still the keystone in the diagnosis of Muir-Torre syndrome.
References
1. Muir EG, Bell AJY, Barlow KA. Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. Br J Surg. 1967; 54:191-5. [PubMed]2. Torre D. Multiple sebaceous tumors. Arch Dermatol. 1968;98:549-51. [PubMed]
3. Reiffers J, Laugier P, Hunziker N. Hyperplasies sébacées, kérato-acanthomes, épithéliomas de visage et cancer du côlon: une nouvelle entité? Dermatologica. 1976;153:23-33. [PubMed]
4. Lynch HT, Lynch PM, Pester J, Fusaro RM. The cancer family syndrome: rare cutaneous phenotypic linkage of Torre's syndrome. Arch Intern Med. 1981;141:607-611. [PubMed]
5. Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33(1):90-104. [PubMed]
6. Kruse R, Lamberti C, Wang Y, Ruelfs C et al. Is the mismatch repair deficiency type of Muir-Torre syndrome confined to mutations in the hMSH2 gene? Hum Genet. 1996;98:747-750. [PubMed]
7. Bapat B, Xia L, Madlensky L, Mitri A et al. The genetic basis of Muir-Torre syndrome includes the hMLH1 locus. Am J Hum Genet. 1996;59:736-739. [PubMed ]
8. Cohen PR, Kohn SR, Davis DA, Kurzrock R. Muir-Torre syndrome. Dermatol Clin. 1995;13:79-89. [PubMed]
9. Ponti G, Ponz de Leon M. Muir-Torre syndrome. Lancet Oncol. 2005;6:980-987. [PubMed]
10. Edelmann W, Yang K, Umar A, Heyer J et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;14;91(4):467-77. [PubMed]
11. Berends MJ, Wu Y, Sijmons RH, Mensink RG et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet. 2002;70(1):26-37. [PubMed]
12. Honchel R, Halling KC, Schaid DJ, Pittelkow M et al. Microsatellite instability in Muir-Torre syndrome. Cancer Res. 1994;54:1159-1163. [PubMed]
13. Mathiak M, Rütten A, Mangold E, Fischer HP et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of imunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343. [PubMed]
14. Ponti G, Losi L, Di Gregorio C, Roncucci L et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, micosatellite instability and immunohistochemistry. Cancer. 2005;103(5):1018-25. [PubMed]
15. Mangold E, Pagenstecher C, Leister M, Mathiak M et al. A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet. 2004;41:567-572. [PubMed]
16. Ponti G, Ponz de Leon M, Losi L, Di Gregorio C et al. Different phenotypes in Muir-Torre syndrome: clinical and biomolecular characterization in two Italian families. Br J Dermatol. 2005;152:1335-38. [PubMed]
17. Harwood CA, Swale VJ, Bataille VA, Quinn AG et al. An association between sebaceous carcinoma and microsatellite instability in immunosuppressed organ transplant recipients. J Invest Dermatol. 2001;116(2):246-253. [PubMed]
18. Kruse R, Ruzicka T. DNA mismatch repair and the significance of a sebaceous skin tumor for visceral cancer prevention. Trends Mol Med. 2004;10(3):136-41. [PubMed]
19. Kruse R, Rütten A, Hosseiny-Malayeri HR, Bisceglia M et al. "Second hit" in sebaceous tumor from Muir-Torre patients with germline mutations in MSH2: allele loss in not the preferred mode of inactivation. J Invest Dermatol. 2001;116(3):463-65. [PubMed]
20. Kruse R, Rütten A, Schweiger N, Jakob E et al. Frequency of microsatellite instability in unselected sebaceous gland neoplasias and hyperplasias. J Invest Dermatol. 2003;120(5): 858-864. [PubMed]
21. Piñol V, Castells A, Andreu M, Castellví-Bel S et al. Accuracy of revised Bethesda guidelines, microsatellite instability and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293(16):1986-94. [PubMed]
22. Chialina SG, Fornes C, Landi C, de la Vega Elena CD et al. Microsatellite instability analysis in hereditary non-polyposis colon cancer using the Bethesda consensus panel of microsatellite markers in the absence of proband normal tissue. BMC Med Genet. 2006;7:5. [PubMed]
23. Fiorentino DF, Nguyen JC, Egbert BM, Swetter SM et al. Muir-Torre syndrome: confirmation of diagnosis by immunohistochemical analysis of cutaneous lesions. J Am Acad Dermatol. 2004;50(3):476-8. [PubMed]
24. Hampel H, Frankel W, Martin E, Arnold M et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18)1851-1860. [PubMed]
25. Wijnen J, de Leeuw W, Vasen H, van der Klift H et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23:142-144. [PubMed]
26. Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271-272. [PubMed]
27. Suchy J, Kurzawski G, Jakubowska A, Lubiński J. Ovarian cancer of endometrial type as part of the MSH6 gene mutation phenotype. J Hum Genet. 2002;47:529-31. [PubMed]
28. Wagner A, Hendriks Y, Meijers-Heijboer EJ, de Leeuw WJF et al. Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet. 2001;38:318-322. [PubMed]
29. Barnetson RA, Tenesa A, Farrington SM, Nicholl ID et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354(26):2751-63. [PubMed]
30. Graells J, Marcoval A, Badell A, Notario J et al. Muir-Torré syndrome in a patient with acquired immunodeficiency syndrome. Br J Dermatol. 1996;135:159-161. [PubMed]
© 2008 Dermatology Online Journal