Eosinophilic fasciitis/generalized morphea overlap
Published Web Location
https://doi.org/10.5070/D36148k6d0Main Content
Eosinophilic fasciitis/generalized morphea overlap
Noushin Heidary MD, Wang Cheung MD, Nadia Wang MD, Hideko Kamino MD, Andrew G Franks Jr MD
Dermatology Online Journal 15 (8): 2
Department of Dermatology, New York UniversityAbstract
A 50-year-old woman presented with a three-month history of violaceous, non-tender, indurated plaques on the chest, abdomen, breasts, and proximal portions of the arms and legs. An incisional biopsy specimen showed changes consistent with a diagnosis of inflammatory morphea. Over the course of one year, the patient began to develop signs and symptoms suggestive of a diagnosis of eosinophilic fasciitis, which included the characteristic groove sign on the upper extremities. Although our patient did not exhibit peripheral or histopathologic evidence of eosinophilia, the diagnosis of eosinophilic fasciitis could still be made because the aforementioned phenomena are not required for diagnosis. Multitude treatment regimes have been reported in the literature as single case reports or small patient series. Our patient was maintained on methrotrexate, oral glucocorticoids, and etanercept with improvement of skin lesions and mobility.
History
 |  |
| Figure 1 | Figure 2 |
|---|---|
A 50-year-old woman with a past medical history of gastroesophageal reflux disease, uterine fibroids, and herpes zoster initially presented to the Charles C. Harris Skin and Cancer Pavilion in November, 2007, with a three-month history of indurated, non-tender lesions on her chest, abdomen, back, and thighs. The initial lesions on the right thigh were targetoid- appearing. An initial evaluation performed by an outside dermatologist showed a normal western blot analysis for Lyme borreliosis and a biopsy specimen suggestive of lupus panniculitis. The patient sequently was referred to a rheumatologist for further evaluation of presumed lupus erythematosus; extensive serum studies showed no abnormalities. Before presenting to the Charles C. Harris Skin and Cancer Pavilion, she was treated with topical glucocorticoids and intralesional triamcinolone without improvement. The use of oral glucocorticoids as monotherapy resulted in improvement of the skin lesions but were discontinued because of diaphoresis and palpitations.
The patient reported a chronic cough and the new-onset arthritis in the knees and hips. She denied recent travel, new medication use, fevers, chills, malaise, Raynaud phenomenon, visual changes, headaches, or shortness of breath. The patient reported edema of the lower extremities that was not alleviated with compression stockings. Medications included occasional aspirin and over-the-counter cough preparations. Family history included a sister with rheumatoid arthritis. An incisional biopsy specimen was obtained from one of the tender plaques on the thigh.
The patient reported worsening of disease activity on hydroxychloroquine, and this medication was subsequently discontinued. The patient is currently maintained on etanercept, methotrexate, and oral glucocorticoids, with a beneficial clinical response.
Physical examination
Erythematous, violaceous, indurated, non-tender plaques without overlying surface changes were present on the chest, abdomen, breasts, proximal arms, and legs, with sparing of the face, hands, and feet. Prominence of the venous vasculature of the forearms was noted. There was 1+ edema of the lower extremities.
Laboratory data
A complete blood count with differential analysis, comprehensive metabolic panel; serum protein electrophoresis; angiotensin converting enzyme; SS-A, SS-B, antinuclear, proteinase-3, myeloperoxidase, and anti-ds DNA antibodies; rheumatoid factor; ribonuclear protein; and 24-hour urine heavy metal quantitative levels for arsenic, lead, mercury, thallium, cadmium, and cobalt were normal or negative. A chest radiograph showed no abnormalities.
Histopathology
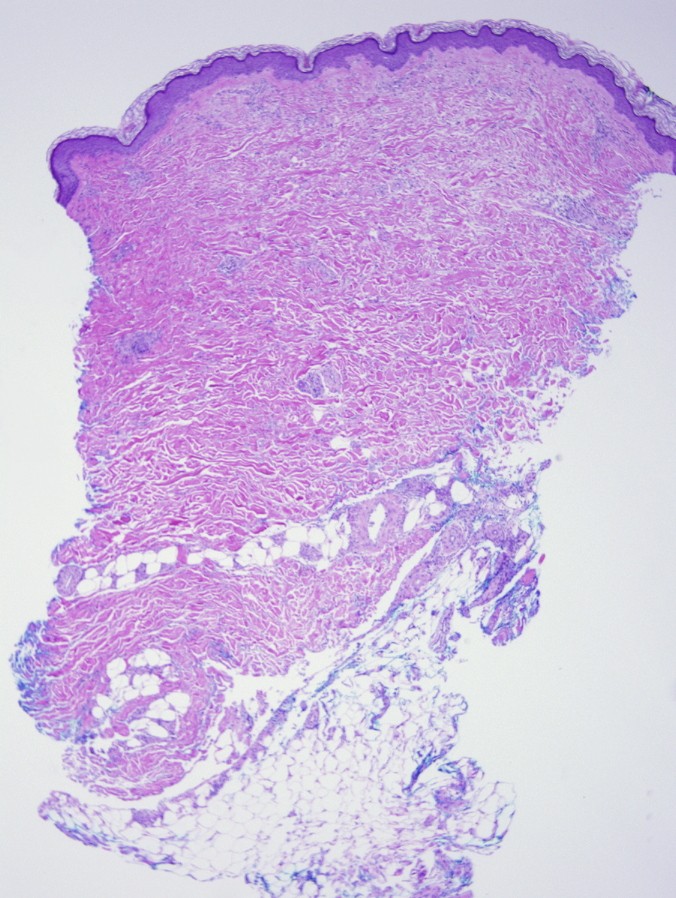
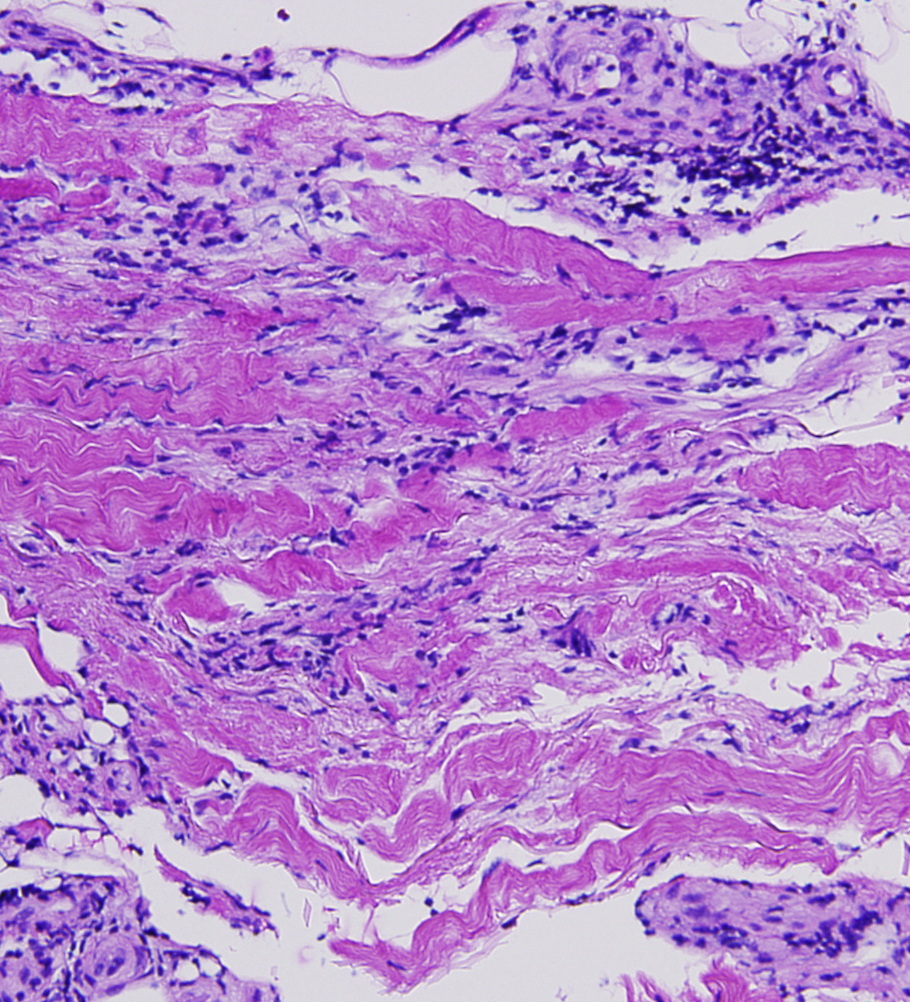
There is a superficial and deep, perivascular and slightly interstitial infiltrate that is comprised of lymphocytes and plasma cells. The infiltrate extends into the subcutaneous septae and the superficial fascia. There are thick collagen bundles in the dermis and subcutaneous septae, with mildly thick superficial fascia. Verhoeff-van Gieson stain highlights some elastic fibers compressed by thick collagen bundles. CD34 shows markedly decreased positive dermal dendritic cells in the reticular dermis and subcutaneous septae.
Comment
Eosinophilic fasciitis (EF) is a scleroderma-like syndrome that was first described in 1974 by Shulman in patients with diffuse fasciitis and eosinophilia. It is a rare disorder with variable clinical manifestations and with overlapping features of other diseases, such as deep morphea and scleroderma. Therefore, its diagnosis may be delayed. The discussion of whether EF is a variant of morphea or a distinct entity is still ongoing. Eosinophilic fasciitis that developed after localized scleroderma has been reported, which suggests a close relationship between the two entities [1]. Although the initial biopsy was suggestive of morphea, the clinical progression of the disease in our patient reflects a diagnosis of eosinophilic fasciitis.
Eosinophilic fasciitis typically affects adults in the second to sixth decades of life although pediatric cases have been described [2, 3]. The etiology of EF is unclear. Reports of extreme physical exercise as well as ingestion of certain pharmaceutical agents, such as statins and L-tryptophan, have been implicated in initiating the onset of this disease [1]. Other potential triggers that have been suggested include trauma, arthropod bites, Lyme borreliosis, hematologic malignant conditions [4], and thyroid disease [5].
The pathogenesis of EF remains unknown. An aberrant immune response has been proposed, which is supported by the findings of hypergammaglobulinemia in the peripheral blood and IgG and C3 deposition in the fascia of some patients [6]. Patients with EF also demonstrate high levels of metalloproteinase I (TIMP-1), which is a protein enzyme that is often elevated in patients with an autoimmune propensity [7]. Kahari et al. reported elevated transforming growth factor B, type I procollagen, and cellular fibronectin mRNA in fibroblasts cultured from lesional tissue [8].
The classic clinical manifestation of EF is a symmetrical, scleroderma-like thickening of the skin of the limbs [9]. The trunk and neck can be affected, with typical sparing of the face and hands. Most patients start with an edematous phase that is accompanied by pitting edema of the extremities. The medial aspects of the upper extremities typically show the groove sign or an indentation along the course of the superficial veins. The physical finding is likely due to the relative sparing of the epidermis and superficial dermis by the fibrotic process coupled with relative mobility of the connective tissue around the remainder of the veins. Thus the superficial layers of skin can bow inward as the peripheral venous pressure falls.
Lesions of morphea on different parts of the body are present in 30 percent of cases [1]. Extracutaneous involvement most commonly presents as synovitis, arthritis, contractures, or carpal tunnel syndrome. Lack of sclerodactyly, Raynaud phenomenon, and visceral involvement helps to distinguish this entity from scleroderma.
Hypergammaglobulinemia, peripheral eosinophilia, and an increased erythrocyte sedimentation rate are features of EF, but normal levels do not rule out the diagnosis. Although eosinophilia may be seen on histopathologic examination of the fascia (often in association with a peripheral eosinophilia), its presence is not required for the diagnosis. Antinuclear antibodies and SSc-specific autoantibodies usually are absent [9].
Although spontaneous recovery is possible in EF, more frequently the signs and symptoms persist and require therapy. Most patients respond to glucocorticoids, but no single treatment works reliably. Cyclosporine, methotrexate, cimetidine, hydroxychloroquine, chloroquine, penicillamine, azathioprine, griseofulvin, ketotifen, and both psoralen-UVA and extracorporeal photochemotherapy have been used with limited evidence [10]. Infliximab was used successfully in conjunction with glucocorticoids and methotrexate in a juvenile eosinophilic fasciitis patient [11]. Dapsone has been described in a single case report to work synergistically with oral glucocorticoids to enhance clinical response [10].
References
1. Antic M, et al. Eosinophilic fasciitis 30 years after-what do we really know? Dermatology 2006; 213: 93 [PubMed]2. Quintero-Del-Rio AI, et al. Faces of eosinophilic fasciitis in childhood. J Clin Rheumatol 2002; 8: 99 [PubMed]
3. Ortega-Loayza AG, et al. Eosinophilic fasciitis in a female child. J Am Acad Dermatol 2008; 58: S72 [PubMed]
4. Kim H, et al. Eosinophilic fasciitis preceding relapse of peripheral T-cell lymphoma. J Korean Med Sci 2000; 15: 346 [PubMed]
5. Farrell AM, et al. Eosinophilic fasciitis associated with autoimmune thyroid disease and myelodysplasia treated with pulsed methylprednisolone and antihistamines. Br J Dermatol 1999; 140: 1185 [PubMed]
6. Moore TL, Zuckner J. Eosinophilic fasciitis. Semin Arthritis Rheum 1980; 9: 228 [PubMed]
7. Jinnin M, et al. Serum levels of tissue inhibitor of metalloproteinase 1 and 2 in patients with eosinophilic fasciitis. Br J Dermatol 2004; 151: 407 [PubMed]
8. Kahari VM, et al. Eosinophilic fasciitis: increased collagen production and type I procollagen messenger RNA levels in fibroblasts cultured from involved skin. Arch Dermatol 1990; 126: 613 [PubMed]
9. Bielsa I, et al. Deep morphea. Semin Cutan Med Surg 2007; 26: 90 [PubMed]
10. Smith L, et al. Dapsone treatment for eosinophilic fasciitis. Arch Dermatol 2008; 144: 845 [PubMed]
11. Tzaribachev N, et al. Infliximab effective in steroid-dependent juvenile eosinophilic fasciitis. Rheumatology 2008; 47: 930 [PubMed]
© 2009 Dermatology Online Journal