A family with Fabry disease diagnosed by a single angiokeratoma
Published Web Location
https://doi.org/10.5070/D35bk233zqMain Content
A family with Fabry disease diagnosed by a single angiokeratoma
Andrea Corry1, Cliona Feighery1, David Alderdice2, Fiona Stewart3, Maureen Walsh4, Olivia M Dolan1
Dermatology Online Journal 17 (4): 5
1. Department of Dermatology, Royal Victoria Hospital, Belfast Health and Social Care Trust, Belfast, Ireland2. Department of Dermatology, Causeway Hospital, Coleraine, Ireland
3. Department of Genetics, Belfast City Hospital, Belfast Health and Social Care Trust, Belfast, Ireland
4. Department of Pathology, Royal Victoria Hospital, Belfast Health and Social Care Trust, Belfast, Ireland
Abstract
This case presents a 39-year-old gentleman with a single angiokeratoma on the abdomen. Because of a family history of early onset cardiac disease, testing for Fabry disease was performed and a mis-sense mutation (A143T) in the Fabry gene confirmed the diagnosis. The unusual aspect of this case is that the patient otherwise had normal health. His only detectable abnormality was a high serum creatinine at 116 mmol/L. Two further affected males and four carrier females were detected on family screening. We tested a further five patients with a single angiokeratoma for Fabry disease. In the five tested though, no suggestive personal or family history was given for any of the patients and no further cases were detected. This case highlights the need for vigilance within dermatology clinics to consider Fabry disease even if a solitary angiokeratoma is the only presenting feature. Some patients do display a milder phenotype and thus a detailed family history should always be taken. As in this case, a solitary angiokeratoma and a suspicious family history may be the only clue. Because enzyme replacement therapy is now available, the potential benefits for the patient and their family are high.
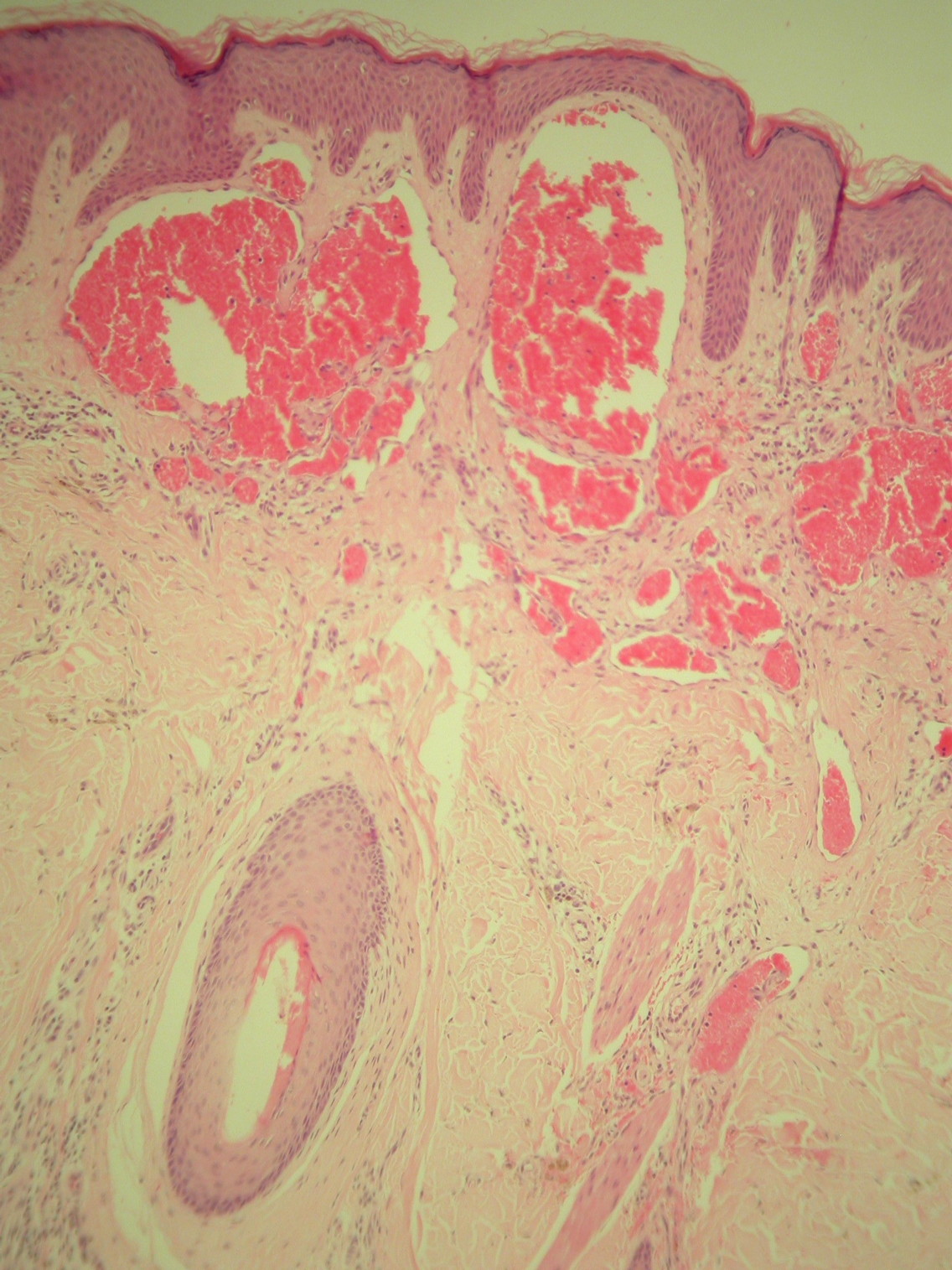 |
| Figure 1 |
|---|
This case presents a 39-year-old gentleman with a single angiokeratoma (Figure 1) excised from the abdomen. Because of a family history of cardiac disease, testing for Fabry disease was performed. The serum α-galactosidase A level was low at 1.5 µmol/L (normal 3-20) and a mis-sense mutation (A143T) was present in the α-galactosidase gene, confirming the diagnosis.
The surprising feature of this case is that the patient exhibited no other physical signs of Fabry disease. He had no history of acroparesthesia and his health was otherwise normal. Cardiac investigations, ophthalmic and neurological examination, and MRI brain were normal. His only abnormality was a slightly low estimated Glomerular Filtration Rate (eGFR) of 70 mL/min.
Genetic screening of the maternal family detected a further two affected males (an asymptomatic middle-aged cousin, and an uncle with early onset cardiac disease), and four carrier females (the patient’s two asymptomatic young daughters, his mother with late onset epilepsy, and an aunt with early onset cardiac disease). No further relevant family history could be elicited.
We retrospectively examined our records for other patients recently seen with an angiokeratoma and the local ethics committee approved testing for Fabry disease. Each had a detailed personal and family history recorded and their serum α-galactosidase A level tested. In the five tested no suggestive personal or family history was given and no further cases of Fabry disease were detected.
Discussion
Fabry disease is an X-linked storage disease caused by deficiency of the enzyme α-galactosidase A. A spectrum of clinical manifestations are seen, with angiokeratoma (Figure 1) being considered the cutaneous hallmark; 66 percent of males and 36 percent of females exhibit at least one [1]. However, the diagnosis of Fabry disease is often delayed until after end organ damage has occurred, with the mean age of death in men being 50 [2].
Previously, atypical cases have been reported associated with mis-sense mutations such as N215S and A143T [1]. Typically these patients have some residual enzyme activity and thus a milder spectrum of disease, usually with no skin involvement and a later onset of disease. In fact, dermatological involvement in Fabry disease has often been considered to be a marker for more severe disease [1], but as this patient illustrates this is not always the case, and milder atypical cases can indeed present with cutaneous features.
The incidence of Fabry disease has been reported to be 1 in 117,000 [3], although recent neonatal screening programs suggest the actual level may be as high as 1 in 3100 [4], possibly reflecting a higher than anticipated incidence of atypical cases. In 2001, enzyme replacement therapy became available and has since been proven to have a beneficial effect on pain, quality of life, and renal and cardiac manifestations [1].
Serum α-galactosidase A levels can now be very easily checked on a dried blood spot. In males demonstration of a low serum α-galactosidase A level is sufficient for diagnosis, but in females genetic studies are mandatory because their α-galactosidase A level may be normal. A database of phenotypes and genotypes in Fabry disease has recently been published [5].
This case highlights the need for vigilance within dermatology clinics to consider Fabry disease even if a solitary angiokeratoma is the only presenting feature. Some patients with mis-sense mutations do display a milder phenotype with late onset [1] and thus a detailed family history should always be taken. As in this case, a solitary angiokeratoma and a suspicious family history may be the only clues, but with availability of enzyme replacement therapy the potential benefits for the patient and family are high.
References
1. Orteu CH, Jansen T, Lidove O, Jaussaud R, Hughes DA, Pintos-Morell G, Ramaswami U, Parini R, Sunder-Plassman G, Beck M, Mehta AB; FOS Investigators. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. 2007 Aug;157(2):331-7. [PubMed]2. MacDermott KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001 Nov;38(11):750-60. [PubMed]
3. Meikle PJ, Hopwood JJ, Clague AE, Carey WF: Prevalence of lysosomal storage disorders. JAMA. 1999 Jan 20;281(3):249-54. [PubMed]
4. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006 Jul;79(1):31-40. [PubMed]
5. Saito S, Ohno K, Sakuraba H. Fabry-database.org: database of the clinical phenotypes, genotypes and mutant α-galactosidase A structures in Fabry disease. J Hum Genet. 2011 Mar 17. [PubMed]
© 2011 Dermatology Online Journal