Cutaneous Rosai-Dorfman disease
Published Web Location
https://doi.org/10.5070/D33sb4b9s4Main Content
Cutaneous Rosai-Dorfman disease
Melissa A Rubenstein MD, Neil N Farnsworth MD, Josie A Pielop MD, Ida F Orengo MD, Jonathan L Curry MD, Carol R Drucker MD,
Sylvia Hsu MD
Dermatology Online Journal 12 (1): 8
Department of Dermatology, Baylor College of Medicine. shsu@bcm.eduAbstract
Sinus histiocytosis with massive lymphadenopathy, or Rosai-Dorfman disease, is a benign idiopathic histiocytic proliferative disorder that commonly involves the lymph nodes but secondarily may involve the skin. However, purely cutaneous disease without lymphadenopathy or internal organ involvement rarely may occur. We present case reports of three patients who presented with asymptomatic nonspecific enlarging skin nodules without evidence of lymphadenopathy or internal disease. Histopathologic examination of skin lesions in all patients showed proliferation of large histiocytes with phagocytosed inflammatory cells characteristic of Rosai-Dorfman disease. However, the diagnoses of dermatofibroma, other spindle cell neoplasm, infectious granulomatous process, and other xanthohistiocytic proliferations were also considered due to the presence of storiform spindle cells and foamy cells in the first case. One patient experienced regression during a course of oral steroids, while another patient cleared spontaneously. In the absence of massive lymphadenopathy characteristic of Rosai-Dorfman disease, the diagnosis of purely cutaneous Rosai-Dorfman disease may be complicated by the rarity, non-specific clinical appearance of skin lesions, and broad histopathological differential diagnosis of this disorder. A high index of suspicion of the clinician and pathologist is often required.
Introduction
Sinus histiocytosis with massive lymphadenopathy (SHML) or Rosai-Dorfman disease (RDD), is a distinct clinicopathological entity first described by Rosai and Dorfman in 1969 [1]. The disease can involve both nodal and extranodal sites, including the skin. It is generally considered a benign, self-limited proliferation of histiocytes. Although cutaneous involvement in RDD is common, purely cutaneous disease is rare. The purely cutaneous form of sinus histiocytosis is referred to as cutaneous RDD rather than SHML, which implies nodal involvement. We report three cases of cutaneous RDD disease.
Case Reports
Case 1
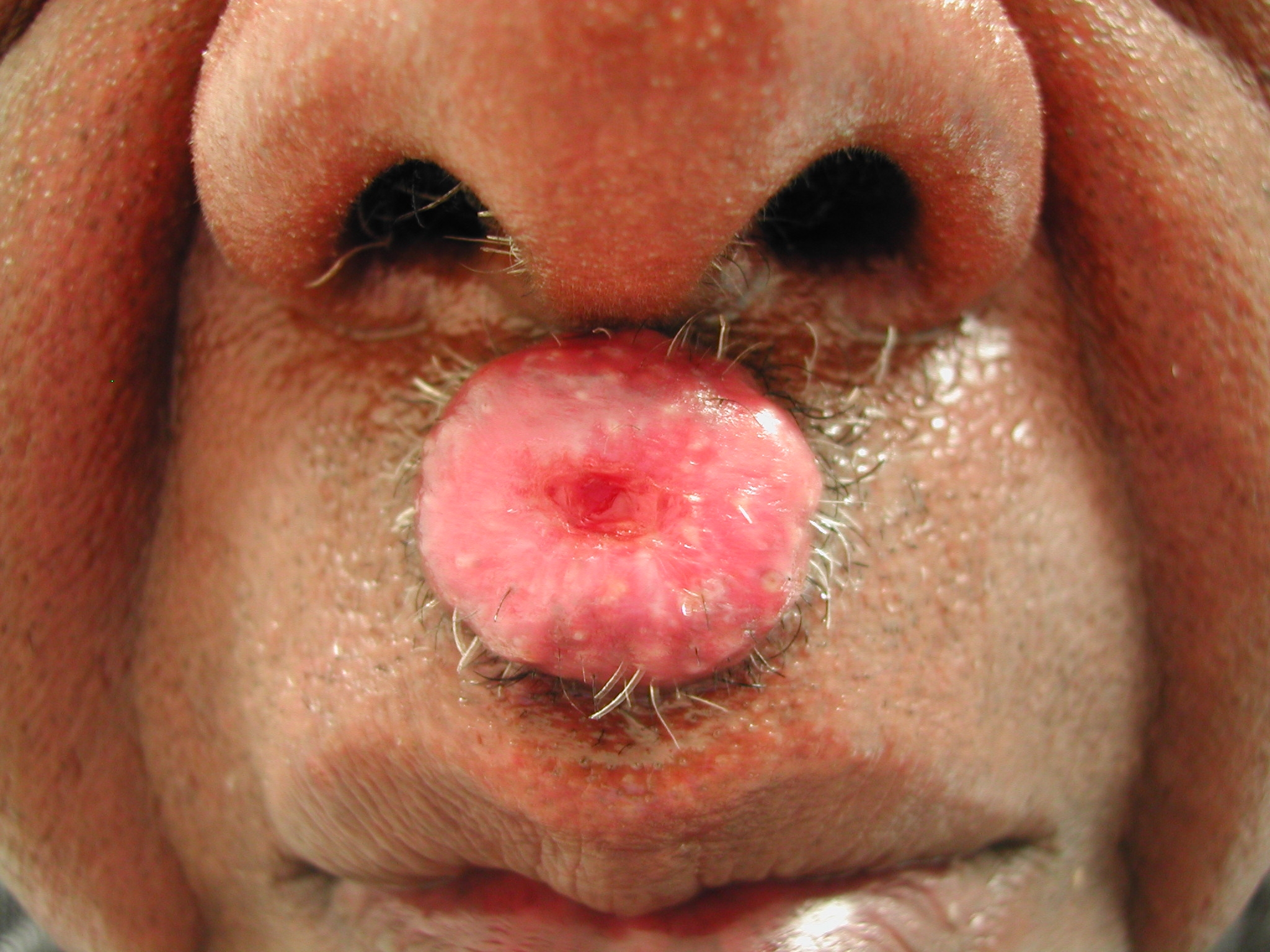
A 65-year-old man with a past medical history of diabetes mellitus, coronary artery disease, osteoarthritis, and hypertension presented with a 2-month history of an asymptomatic lip lesion. Physical examination of the right upper lip revealed a 1.5-cm red-yellow nodule (Fig. 1). The patient had concomitant symptoms of unquantified weight loss, poor appetite, and weakness, but denied fever or night sweats. He had no lymphadenopathy or hepatosplenomegaly. Histological analysis of initial shave biopsy of the lip lesion revealed a dermal proliferation of spindle-shaped, dendritic, and epithelioid cells in a storiform pattern and few cells with foamy cytoplasm thought to be consistent with a cellular benign fibrous histiocytoma or an atypical fibroxanthoma. He underwent Mohs excision of the residual lip lesion requiring a bilateral advancement flap 2 weeks after initial presentation. Histological analysis of Mohs specimen was thought to be most consistent with a spindle cell neoplasm lacking the high-grade atypia of atypical fibroxanthoma. At his post-operative follow-up appointment 1 week later, a 2-cm nodule not present at the time of his Mohs procedure was noted on the right frontal scalp, and a biopsy specimen was obtained. Several consultants interpreted the upper lip and frontal scalp biopsy specimens as a spindle cell neoplasm, non-caseating granulomatous inflammatory process, and diffuse dermatitis secondary to infectious process or unusual xanthomatous process. Multiple special stains including PAS, AFB, Fite, GMS, and Steiner as well as AFB and fungal tissue cultures were negative for microorganisms.
At a follow-up visit 5 weeks after initial presentation, the scalp lesion had enlarged to 4 cm. Tests revealed a normal chest x-ray, and a normal laboratory workup including complete blood count, electrolytes, liver function profile, uric acid, and LDH except for a hemoglobin of 9.8. Further studies showed the anemia to be consistent with an anemia of chronic disease. CT scans of the head, chest, and abdomen were notable only for a soft tissue mass over the right frontal scalp. The patient received oral steroids for 6 weeks during which time the scalp lesion completely resolved. At 9-month followup, the patient remained asymptomatic with no subsequent lymphadenopathy or lesions at other sites.
Repeat histopathological evaluation of all three specimens (shave biopsy of lip, Mohs specimen of lip, and shave biopsy of scalp) revealed a fibrohistiocytic proliferation of spindled cells with multiple foci of superficial acute inflammation in the superficial aspects of the specimens similar to what had previously been described. These superficial cells were CD68 positive, weakly S-100 positive, and negative for lysozyme and CD1a. In addition, deeper sections available from the Mohs specimen of the upper lip revealed clusters of histiocytes with abundant pale cytoplasm and nuclei with vesicular chromatin were observed in the deep dermis and subcutaneous fat. Phagocytosed neutrophils were present in the cytoplasm of some of these cells. These histiocytes were strongly positive for S-100 protein and negative for CD1a, CD68, and lysozyme. The deeper aspects were thought to be strongly suggestive of Rosai-Dorfman disease.
Case 2
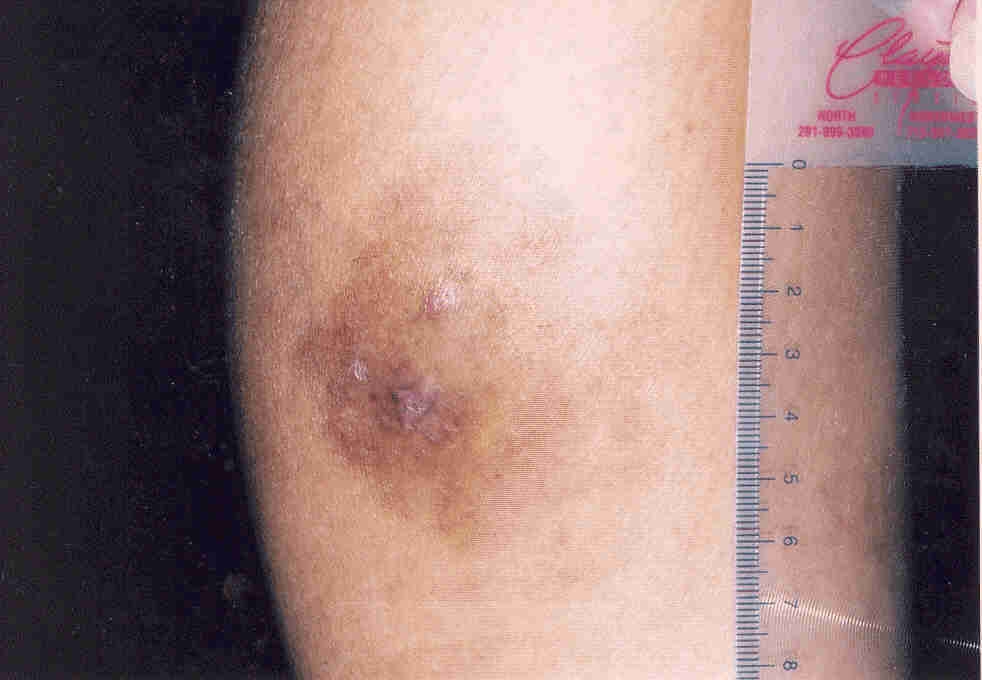
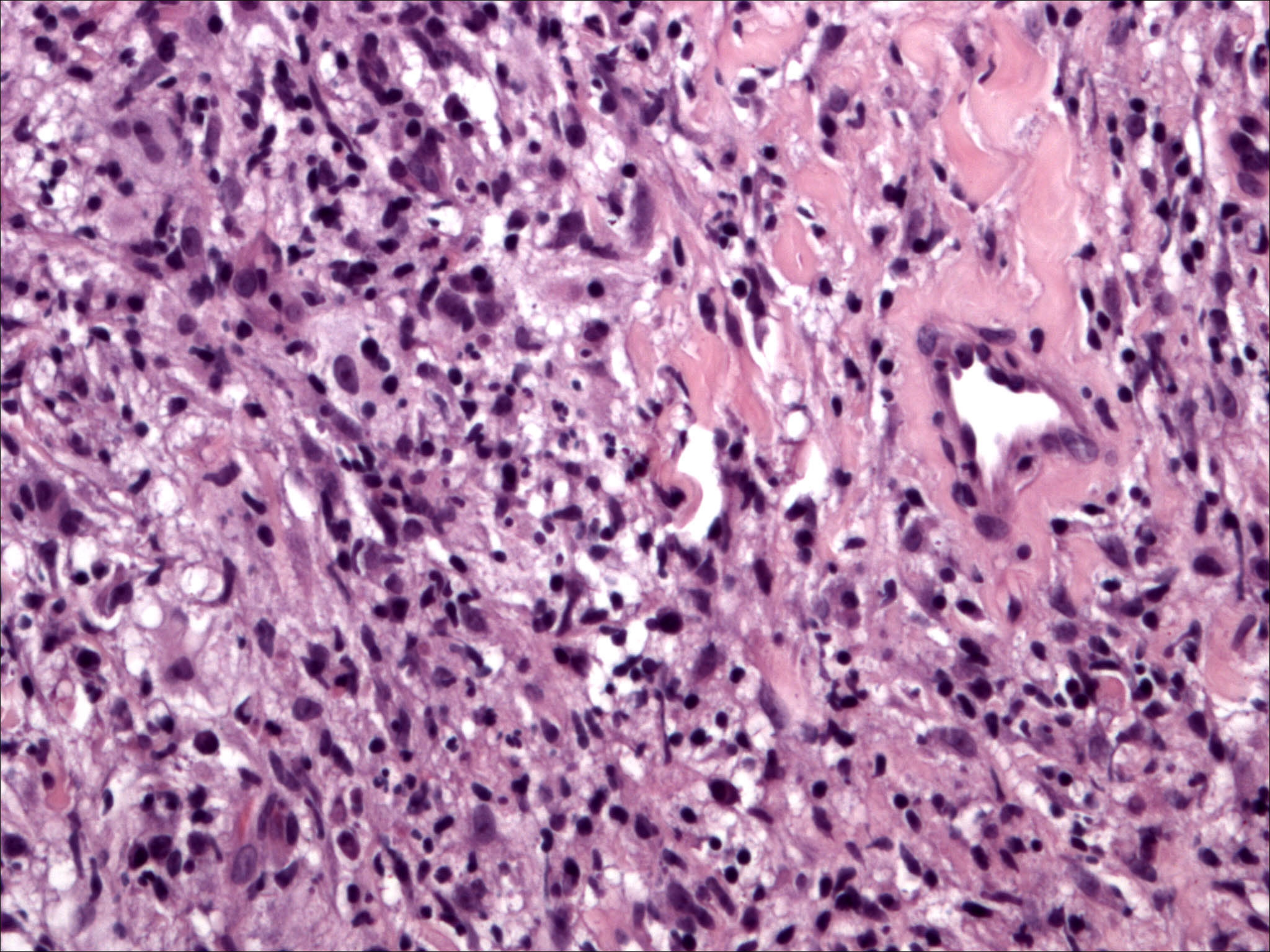
A 43-year-old woman with no past medical history presented with a 2-month history of an enlarging right calf lesion. Examination revealed a 4 × 7 cm violaceous indurated plaque (Fig. 2). The patient had no lymphadenopathy, hepatosplenomegaly, or other associated symptoms. Histologic examination of a biopsy specimen showed uninvolved epidermis separated from the diffuse dermal infiltrate by a thin grenz zone. The infiltrate extended from the papillary dermis to the deep reticular dermis and upper subcutis and was composed of aggregates and sheets of large histiocytic cells with small round-to-oval nuclei and abundant foamy to lightly eosinophilic cytoplasm (Fig. 3). Some cells contained intact lymphocytes. These cells were admixed with a large number of lymphocytes and scattered plasma cells. Special stains were negative for fungal, acid-fast, and bacterial organisms. The large histiocytic cells were strongly positive for S-100 and CD68 protein. The presence of emeripolesis as well as the strong positivity of the histiocytic cells for S-100 protein supported the diagnosis of cutaneous Rosai-Dorfman disease. The lesion resolved without treatment. The patient has remained asymptomatic with no signs of recurrence at 6-month follow-up.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. 1.5 cm red-yellow nodule of right upper lip (case 1) | |
| Figure 2. Violaceous, indurated plaque of right calf (case 2) | |
Case 3
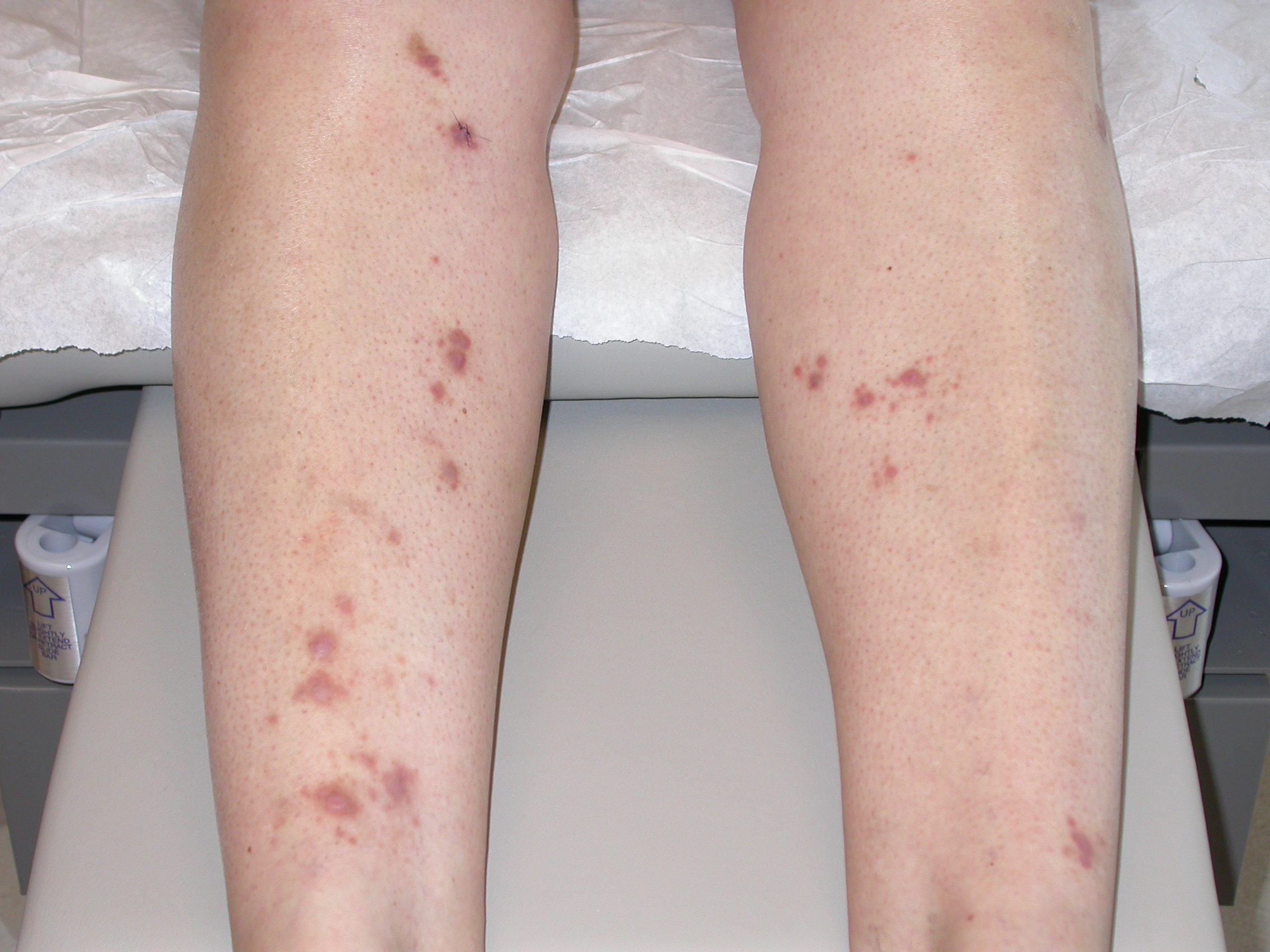
An otherwise healthy 55-year-old woman presented with an 18-month history of nonpruritic lesions on both legs. Examination revealed indurated violaceous papules and nodules over both shins (Fig. 4). The patient had no lymphadenopathy or hepatosplenomegaly. Biopsy specimens from 3 lesions showed nodular dermal aggregates of foamy histiocytes among neutrophils, lymphocytes, and plasma cells. The histiocytic nuclei were uniform, round, and vesicular; phagocytosed plasma cells and lymphocytes could be seen within their abundant, eosinophilic cytoplasm suggestive of Rosai-Dorfman disease.
 |  |
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Aggregates and sheets of large histiocytic cells with small round to oval nuclei and abundant foamy to lightly eosinophilic cytoplasm (H & E 400 ×) (case 2) | |
| Figure 4. Violaceous plaques and nodules of lower extremities (case 3) | |
Comment
Sinus histiocytosis with massive lymphadenopathy (SHML), or Rosai-Dorfman disease (RDD), is a benign histiocytic proliferative disorder clinically characterized by painless cervical lymphadenopathy, fever, leukocytosis, elevated erythrocyte sedimentation rate, and a polyclonal hypergammaglobulinemia. It is more commonly seen in blacks and whites with a slight male predominance and a mean age onset of approximately 20 years [2]. Approximately 40 percnet of patients have extranodal involvement, and although the skin is the most common site, involvement of almost every organ system has been reported [2]. Skin lesions of SHML may be solitary or multiple, macular or papulonodular, xanthomatous or erythematous, dermal or subcutaneous, and found in virtually any location including the face, ears, trunk, extremities, or genetalia [2]. Histologic findings of skin lesion biopsy specimens are similar to those found in lymph nodes, typically being a dermal or subcutaneous dense infiltrate of histiocytes with large vesicular nuclei and abundant pale pink cytoplasm. These cells may contain intracytoplasmic lymphocytes, plasma cells, or neutrophils (emperipolesis). There may be an accompanying inflammatory response consisting of lymphocytes, plasma cells, neutrophils or epithelioid cells. Typically the characteristic histiocytes of RDD are positive for S-100, negative for CD1a, and variably positive for CD68 [3, 4]. Although pathologic evaluation is key to definitive diagnosis, the variable presence of fibrosis[3], vascular proliferation [5], neutrophil microabscesses [3], lymphoid aggregates with germinal centers, and background histiocytic proliferations of foam cells, multinucleated giant cells, and/or Touton cells [5, 6] may result in confusion of this disorder with a neoplastic, xanthomatous, infectious, or other histiocytic process. Given the wide range of clinical presentations and the broad pathologic differential diagnosis, the clinical hallmark of massive lymphadenopathy is often crucial for diagnosis of SHML.
Cutaneous findings of RDD may be present without lymph node involvement. Although SHML itself is relatively rare, purely cutaneous RDD is even less common, accounting for approximately 3 percent of SHML cases in one large series [2]. There are no distinguishing clinical or histologic features of skin lesions in SHML to differentiate them from the skin lesions of purely cutaneous RDD [5, 7, 8]. Cutaneous RDD findings are nonspecific and may present as single or multiple macules, papules, plaques, or nodules ranging in color from red-brown to orange to yellow [3, 7, 9]. The lesions may be indurated and can show central atrophy or ulceration [2, 7, 10]. The lesions of RDD show no predilection for any one location and can localize in the soft tissue mimicking a subcutaneous mass or panniculitis [7, 11, 12, 13]. Cutaneous RDD has been clinically mistaken for other dermatologic disorders including vasculitis [14], acne vulgaris [15], hidradenitis suppurativa [6], granuloma annulare [16], lupus vulgaris [15], sarcoidosis [15], malignant breast neoplasm [17], and other histiocytoses [15].
Several authors suggest that cutaneous RDD is a distinct clinical entity because of its unique epidemiology and lack of systemic involvement even with long-term followup. Patients with cutaneous RDD seem to have an older age of onset of disease (43.5 in one series) and higher rates of disease in women, Asians and whites [3, 7, 12]. Most patients with cutaneous RDD do not have associated systemic symptoms such as fever, malaise or night sweats, although a few have been reported to have fatigue and weight loss similar to our findings in case 1 [15]. RDD has been reported in association with nonspecific laboratory findings including mild anemia (case 1) [3, 7, 18], elevated ESR [3, 15], and polyclonal hypergammaglobulinemia [4, 7, 16, 20], and an association with immune-mediated diseases such as hypothyroidism [19], lymphoma [20], and lupus erythematosus [3]. Cutaneous RDD patients may have other sites of extranodal disease, most commonly uveitis [3, 7, 19, 20].
Although histopathological examination is often the key to diagnosis, especially in the absence of lymphadenopathy or other systemic symptoms, it is worth noting that the difficulty in obtaining a pathological diagnosis we experienced in case 1 is not unusual in cases of purely cutaneous RDD. Several reported cases were initially thought to be consistent with a xanthomatous process such as juvenile xanthogranuloma or tuberous xanthoma [6, 10, 17], histiocytic disorder [6], neoplastic process including giant cell tumor of tendon sheath [16], and epithelioid sarcoma [17], and infectious or other inflammatory disorder [8, 21], including granuloma annulare [16], inflammatory pseudotumor [20], granulomatous mastitis [17], and panniculitis of unknown cause [13]. Other diseases often included in the pathological differential diagnosis of RDD include Langerhans cell histiocytosis, malignant histiocytosis, hemophagocytic syndrome, reticulohistiocytoma, Tangier disease, dermatofibroma, and lymphoma [5, 9]. Our first case was described as consistent with a cellular benign fibrous histiocytoma (dermatofibroma), spindle cell neoplasm, inflammatory granulomatous process, dermatitis secondary to an infectious process, and a xanthomatous process before deeper sections revealed a histiocytic proliferation more typical of RDD. Several pitfalls may complicate pathologic diagnosis. First, variable amounts of fibrosis may be present in cutaneous lesions of RDD [3, 10]. When present in a storiform pattern, confusion with a dermatofibroma may occur [3], as in our case, although the cytoplasm of the histiocytes is not as abundant as in RDD and emperipolesis is absent [5]. In addition, there may be a background histiocytic proliferation of foam cells, multinucleated giant cells, or Touton cells leading to confusion with other xanthohistiocytic disorders [3, 5, 6, 10] Fibrosis and foamy histiocytes may be especially prominent in older lesions.10 Finally, epidermal changes including pseudoepitheliomatous hyperplasia may be present [8, 20]. Tissue cultures, special stains for infection, immunohistochemical studies, and possibly even electron microscopy to evaluate for Birbeck granules may be required before the diagnosis of RDD is reached.
It is conceivable that some patients with purely cutaneous symptoms may have clinically undetectable systemic lesions. However, there have been no reported cases of cutaneous disease developing into systemic disease. The clinical course is variable, with some lesions resolving spontaneously over weeks to months [7, 8, 12, 23, 14] and others persisting for years [7, 10, 15, 16] or even recurring after excision [6]. Treatment is generally not necessary for cutaneous RDD, but may be desired for cosmetic purposes or symptomatic relief. Cutaneous lesions have been reported to respond to radiotherapy[3, 6], cryotherapy [15, 18], excision [3, 4, 7]] topical[4], and oral corticosteroids (case 1) [3, 19], and high-dose thalidomide [7, 22]. Investigation of the true efficacy of any of these therapeutic modalities is complicated by the rarity of this disorder.
RDD should be considered in patients with histiocytic like papules, pustules, nodules, or plaques without any nodal or organ enlargement. Despite similar histopathological features in both nodal and extranodal sites, a diagnosis of cutaneous RDD is often overlooked in the absence of lymphadenopathy. Infrequent occurrence of cutaneous RDD, lack of any distinguishing clinical features of cutaneous lesions, and a broad histologic pathologic differential diagnosis may complicate diagnosis.
References
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.2. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Pitamber HV, Grayson W. Five cases of cutaneous Rosai-Dorfman disease. Clin Exp Dermatol.
5. Thawerani H, Sanchez RL, Rosai J, Dorfman RF. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
6. Lazar AP, Esterly NB, Gonzalez-Crussi F. Sinus histiocytosis clinically limited to the skin. Pediatr Dermatol. 1987;4:247-53.
7. Lu C, Kuo T, Wong W, Hong H. Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman disease in Taiwan. J Am Acad Dermatol 2004;51:931-9.
8. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
9. Perrin C, Michiels JF, Lacour JP, Chagnon A, Fuzibet JG. Sinus histiocytosis (Rosai-Dorfman disease) clinically limited to the skin. An immunohistochemical and ultrastructural study. J Cutan Pathol. 1993;20:368-374.
10. Quaglino P, Tomasini C, Novelli M, Colonna S, Bernengo MG. Immunohistologic findings and adhesion molecule pattern in primary pure cutaneous Rosai-Dorfman disease with xanthomatous features. Am J Dermatopathol. 1998;20:393-398.
11. Puppin D, Jr., Chavaz P, Harms M. Histiocytic lymphophagocytic panniculitis (Rosai-Dorfman disease): a case report. Dermatology. 1992;184:317-320.
12. Skiljo M, Garcia-Lora E, Tercedor J, Massare E, Esquivias J, Garcia-Mellado V. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
13. Suster S, Cartagena N, Cabello-Inchausti B, Robinson MJ. Histiocytic lymphophagocytic panniculitis. An unusual extranodal presentation of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Arch Dermatol. 1988;124:1246-1249.
14. Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
15. Ang P, Tan SH, Ong BH. Cutaneous Rosai-Dorfman disease presenting as pustular and acneiform lesions. J Am Acad Dermatol. 1999;41:335-337.
16. Scheel MM, Rady PL, Tyring SK, Pandya AG. Sinus histiocytosis with massive lymphadenopathy: presentation as giant granuloma annulare and detection of human herpesvirus 6. J Am Acad Dermatol. 1997;37:643-646.
17. Mac-Moune Lai F, Lam WY, Chin CW, Ng WL. Cutaneous Rosai-Dorfman disease presenting as a suspicious breast mass. J Cutan Pathol. 1994;21:377-382.
18. Wang KH, Cheng CJ, Hu CH, Lee WR. Coexistence of localized Langerhans cell histiocytosis and cutaneous Rosai-Dorfman disease. Br J Dermatol. 2002;147:770-774.
19. Salim A, Williamson M, Barker F, Hughes J. Steroid responsive cutaneous Rosai-Dorfman disease associated with uveitis and hypothyroidism. Clin Exp Dermatol. 2002;27:277-279.
20. Kroumpouzos G, Demierre MF. Cutaneous Rosai-Dorfman disease: histopathological presentation as inflammatory pseudotumor. A literature review. Acta Derm Venereol. 2002;82:292-296.
21. Tsang WY, Chan JK, Ho WK, Yu HC, Chow LT. Extranodal Rosai-Dorfman disease: an uncommon cause of persistent nodule in the ear. J Laryngol Otol. 1992;106:249-251.
22. Viraben R, Dupre A, Gorguet B. Pure cutaneous histiocytosis resembling sinus histiocytosis. Clin Exp Dermatol. 1988;13:197-199.
© 2006 Dermatology Online Journal