Mixed immunobullous disorder most consistent with the IgA-form of epidermolysis bullosa acquisita
Published Web Location
https://doi.org/10.5070/D33jr0t3ccMain Content
Mixed immunobullous disorder most consistent with the IgA-form of epidermolysis bullosa acquisita
Carina Rizzo MD, Henry J Votava MD, Shane A Meehan MD, Roopal Kundu MD, Andrew G Franks Jr MD
Dermatology Online Journal 15 (8): 19
Department of Dermatology, New York UniversityAbstract
We describe a case of non-scarring, generalized, cutaneous and mucosal subepidermal bullous dermatosis that is characterized histopathologically by a neutrophilic infiltrate and strong linear staining with both IgA and IgG along the basement-membrane zone. Autoantibodies to collagen VII of both the IgA and IgG4 subtypes were detected by indirect immunofluorescence test, which led led to a diagnosis of epidermolysis bullosa aquisita (EBA). EBA is a subepidermal bullous disorder that is mediated by autoantibodies, which are directed against type VII collagen. The distinct clinical presentations of EBA are reviewed and discussed in the context of the unique autoantibody profile of this case.
 |  |
| Figure 1 | Figure 2 |
|---|
History
A 31-year-old woman was referred to the Charles C. Harris Skin and Cancer Unit in February, 2009, for evaluation of a generalized bullous eruption. The patient had been well until six months prior to presentation when she developed transient, erythematous, pruritic eruptions on the back and chest, which evolved over the course of weeks into spontaneous and mechanically induced vesicles and bullae. The bullous eruption was most pronounced on the neck, chest, arms, and oral mucosa. The face was spared. There was no photosensitivity. There were no antecedent illnesses and no new or changed medications. With the exception of her cutaneous symptoms, the patient denied fever, chills, malaise, arthralgias, myalgias, and neurologic, respiratory, or gastrointestinal symptoms. Past medical history includes renal vein thrombosis two years prior to presentation, which led to a diagnosis of antiphosopholipid antibody syndrome. Warfarin therapy was initiated at that time, and there were no further thrombotic events.
Medications include prednisone 20 mg daily, dapsone 75 mg twice daily, hydroxychloroquine 400 mg daily, cimetidine 400 mg daily, and topical clobetasol ointment. Despite this regimen, the patient continued to develop cutaneous bullae primarily at sites of trauma. She developed painful, perianal erosions and extensive oral erosions that persisted despite a soft diet and rigorous oral care.
Physical Examination
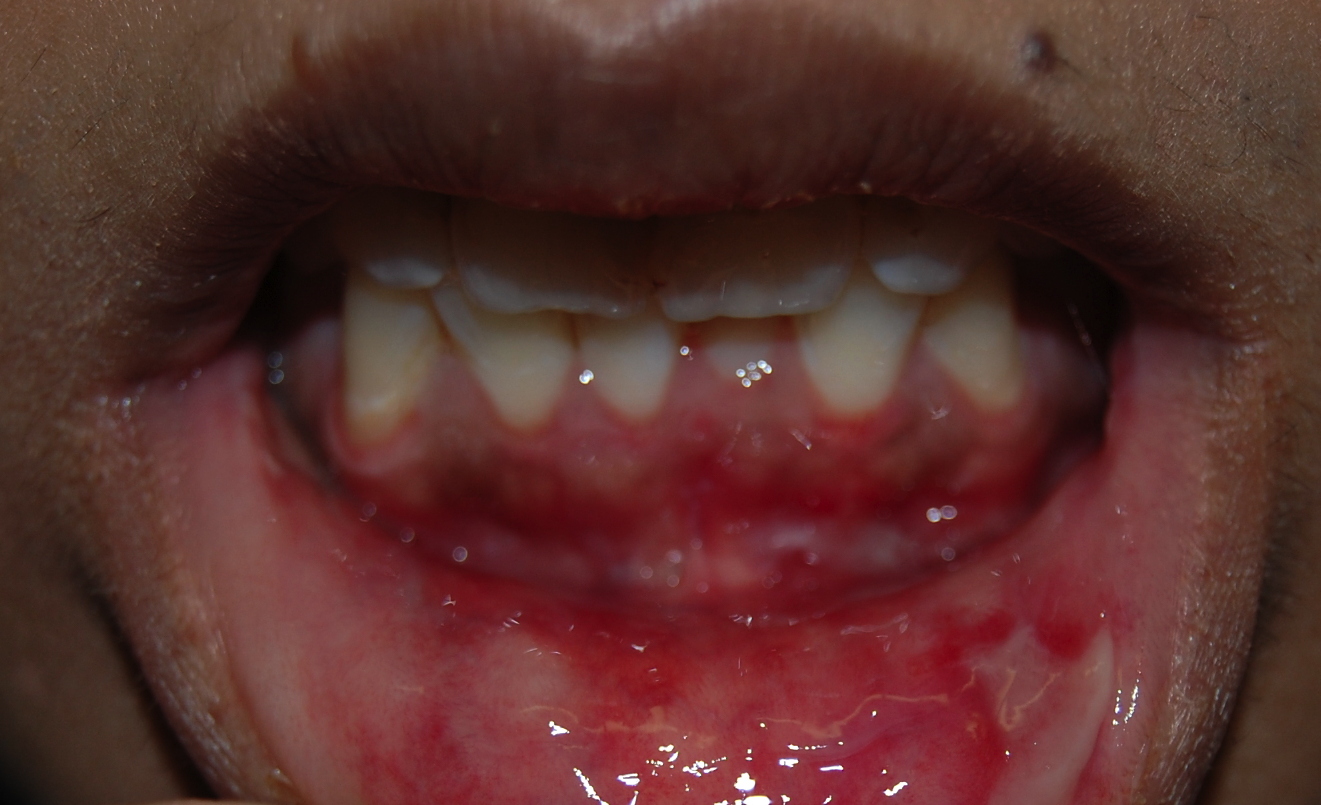
There were hyperpigmented macules and patches as well as grouped vesicles and small tense bullae that were filled with clear fluid on a base of normal skin, which were distributed on the dorsal aspects of the hands, arms, legs, back, and chest. There were extensive erosions of the upper and lower gingival mucosae, the lateral aspect of the tongue, the upper and lower oral labial mucosae, and the anal mucosa. There were no ocular lesions. There was no alopecia, scars, or milia, and the nails were unaffected.
Laboratory data
The hemoglobin was 9.5 gm/dL and hematocrit 28.1 percent. A lupus anticoagulant was positive. The anti-nuclear antibody titer was 1:320, with a homogenous pattern. Double-stranded DNA antibody was detected at 8 IU/ml. Complement levels were normal. Indirect immunofluorescence microscopy using monkey esophagus showed circulating IgG, IgG4, and IgA antibodies against the epidermal basement-membrane zone that localized to the dermal side of salt-split skin. There were no intercellular antibodies detected. Tests on collagen VII deficient skin were negative. Enzyme-linked immunosorbent assay showed antibodies to desmoglein 1 at 21.9 units, desmoglein 3 at 22.9 units, bullous pemphigoid (BP) antigen 180 at 24.9 units and BP antigen 230 at 30.1 units. IgA antiemdomysial antibodies and IgA tissue transglutaminase antibodies were not detected.
Histopathology
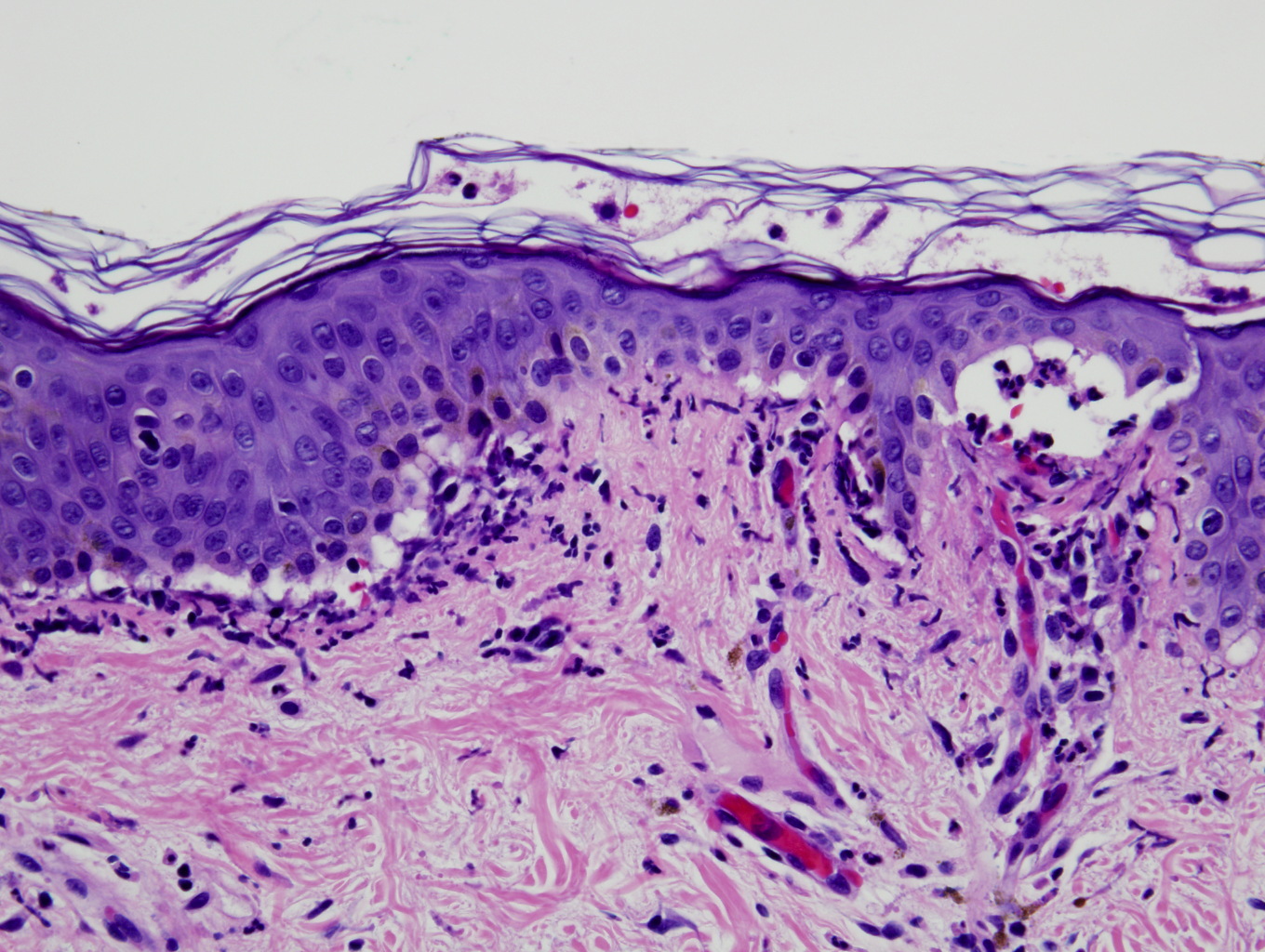
Neutrophils are aligned along the dermoepidermal junction where there are scattered papillary microabscesses and small subepidermal vesicles. There is a superficial, perivascular and interstitial infiltrate comprised of neutrophils and rare eosinophils. A direct immunofluorescence test shows 3+ linear staining of IgG and IgA and 3+ patchy linear staining of IgM, C3, and fibrin below the dermoepidermal junction.
Comment
Epidermolysis bullosa aquisita (EBA) is a subepidermal bullous disorder that is mediated by autoantibodies directed against type VII collagen [1, 2]. It is distinguished immunohistochemically from bullous pemphigoid (BP) by the localization of circulating autoantibodies to the dermal side of salt-split skin, which corresponds to the ultrastructural location of the target antigen.
The classic presentation of EBA is a noninflammatory mechanobullous disease that predominantly affects acral surfaces and sites of trauma and heals with scars and milia, which is reminiscent of hereditary EB. A cicatricial pemphigoid-like presentation has been described, which is characterized by predominantly mucosal erosions that heal with scars [3, 4]. Both conditions are mediated by circulating IgG anti-collagen VII antibodies, which have been demonstrated to be pathogenic by murine passive transfer experiments [2].
The inflammatory variant of EBA overlaps clinically with BP. The typical presentation is of a generalized, pruritic, inflammatory urticarial and bullous eruption [5, 6]. Mucosal involvement may be observed [7]. Similar to the classic variant of EBA, inflammatory EBA also is characterized by circulating IgG anti-collagen VII antibodies. In contrast, however, the inflammatory lesions heal without scars. The molecular and cellular mechanism underlying the difference in clinical presentation has not been elucidated.
The IgA form of EBA is an uncommon subtype that was initially thought to be a variant of linear IGA bullous disease (LABD). Ultrastructural determination linked the pathogenesis to the presence of circulating IgA anti-collagen VII antibodies, which localized to the dermal side of salt-split skin [8, 9]. Histopathologic features include subepidermal blisters with a prominent neutrophilic infiltrate that is sometimes accompanied by eosinophils, which may occur in dermal papilla, a pattern observed in dermatitis herpetiformis (DH) and LABD. A direct immunoflorescence (DIF) test demonstrates linear IgA deposits at the basement-membrane zone (BMZ) [3].
The IgA form of EBA resembles LABD in morphology and a tendency for mucosal involvement [8]. Ocular involvement has been described in several cases [10], but therapy-resistant ocular scars are rare in comparison to classic EBA [8]. Cutaneous and mucosal lesions heal without scars, and there is no nail involvement [11]. In contrast to classic LABD, the IgA form of EBA may be somewhat less responsive to dapsone although others have reported excellent results with this therapy [8].
In bullous systemic lupus erythematosus (SLE), autoimmunity to collagen VII arises in the setting of SLE. Bullous SLE presents with a non-scarring bullous eruption that favors sun-exposed sites. There are DH-like histopathologic features [12], and DIF may show linear or granular IgG, IgM, and IgA at the BMZ.
In this case, the patient fails to meet the American Rheumatism Association criteria for SLE. The absence of clinical scars and strong linear staining with IgA lead us to favor a diagnosis of the IgA-form of EBA, which is based on the spectrum of clinical and immunofluorescence findings. The breadth of the autoantibody profile in this case is unique, however. The low positive detection of antibodies to desmogelin 1 and 3 as well as BP 180 and 230, in the absence of intracellular antibody staining and the absence of deposits on the roof of salt-split skin by IIF, suggest that these are likely epiphenomenon, perhaps arising secondary to epitope spreading in the setting of pronounced cutaneous inflammation. In the limited reports in the literature, IGA-EBA has been defined by the exclusive detection of the IgA antibody subtype to avoid overlap with other defined forms of EBA [8]. A dual IgA and IgG autoimmune response has proved to be common in bullous SLE, BP, and LABD [13] but has not been explored in classic or IgA forms of EBA. In practice, overlap in clinical presentation as well as autoantibody subtype and molecular specificity is common, which poses a diagnostic challenge [14, 15, 16]. In such cases, delineation of the primary target antigen is critical in defining immunobullous disease and interpretation in the context of the clinical presentation in necessary to appropriately direct therapy.
References
1. Woodley DT, et al. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type vii procollagen. J Clin Invest 1988;81:683 [PubMed]2. Woodley DT, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol 2006;126:1323 [PubMed]
3. Woodley DT, et al. Autoimmunity to type VII collagen: epidermolysis bullosa acquisita. Clin Rev Allergy Immunol 2007;33:78 [PubMed]
4. Dahl MG. Epidermolysis bullosa acquisita--a sign of cicatricial pemphigoid? Br J Dermatol 1979;101:475 [PubMed]
5. Briggaman RA, et al. Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol 1985;85:79s [PubMed]
6. Jonkman MF, et al. Inflammatory variant of epidermolysis bullosa acquisita with IgG autoantibodies against type VII collagen and laminin alpha3. Arch Dermatol 2000;136:227 [PubMed]
7. Tokuda Y, et al. A case of an inflammatory variant of epidermolysis bullosa acquisita: Chronic bullous dermatosis associated with nonscarring mucosal blisters and circulating igg anti-type-vii-collagen antibody. Dermatology 1998;197:58 [PubMed]
8. Vodegel RM, et al. Iga-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol 2002;47:919 [PubMed]
9. Woodley DT, et al. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med 1984;310:1007 [PubMed]
10. Bauer JW, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. Br J Dermatol 1999;141:887 [PubMed]
11. Lally A, et al. Dermal-binding linear IgA disease: an uncommon subset of a rare immunobullous disease. Clin Exp Dermatol 2007;32:493 [PubMed]
12. Vassileva S: Bullous systemic lupus erythematosus. Clin Dermatol 2004;22:129 [PubMed]
13. Kromminga A, et al. Patients with bullous pemphigoid and linear iga disease show a dual iga and igg autoimmune response to bp180. J Autoimmun 2000;15:293 [PubMed]
14. Collier P, et al. Molecular overlap of the iga target antigens in the subepidermal blistering diseases. Dermatology 1994;189 (Suppl 1):105 [PubMed]
15. Kirtschig G, et al. Acquired bullous diseases of childhood: re-evaluation of diagnosis by indirect immunofluorescence examination on 1 m NaCl split skin and immunoblotting. Br J Dermatol 1994;130:610 [PubMed]
16. Umemoto N, et al. A case of nonscarring subepidermal blistering disease associated with autoantibodies reactive with both type vii collagen and laminin 5. Dermatology 2003;207:61 [PubMed]
© 2009 Dermatology Online Journal