Tuberous sclerosis
Published Web Location
https://doi.org/10.5070/D331t789d1Main Content
Tuberous sclerosis
Sue Ann Wee MD, Bill Fangman MD
Dermatology Online Journal 13 (1): 22
New York University Department of Dermatology
Abstract
A 46-year-old woman presented with multiple, skin-colored, hyperpigmented, dome-shaped facial papules. Histopathologic examination was consistent with angiofibromas. Clinical history and examination were consistent with tuberous sclerosis. The clinical manifestations, pathogenesis, evaluation, and treatment of tuberous sclerosis are discussed.
Clinical synopsis
A 46-year-old woman presented to the Charles C. Harris Skin and Cancer Pavilion in October 2005 seeking cosmetic treatment of multiple facial papules that had been gradually increasing in number over the past 20 years. Prior diagnoses of her facial papules included acne vulgaris and dermatosis papulosa nigra. Three biopsies from typical facial papules provided the correct diagnosis. The patient underwent bilateral nephrectomy in 2001 for a renal-cell carcinoma (chromophobe-cell type) in the right kidney and glomerulosclerosis and cysts in the left kidney. She also reported a history of seizures. On subsequent clinic visits, it was discovered that her 17-year-old daughter had a diagnosis of tuberous sclerosis. The patient had a normal ophthalmologic examination 2 months ago. The patient also has a history of hypertension and hepatitis-C virus infection.
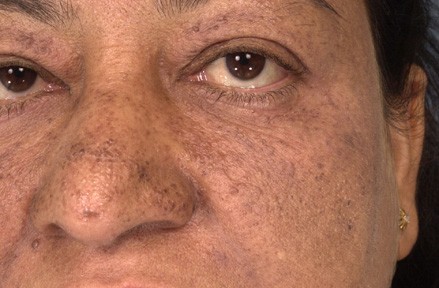
Symmetric hyperpigmentation and numerous, 1-3 mm, skin-colored and hyperpigmented, dome-shaped papules were present on the periorbital regions, cheeks, and nose. Skin-colored, soft plaques with prominent follicular prominence were noted on the right lateral and posterior aspects of the neck and right medial buttock. Firm, flesh-colored papules, some with overlying blue-grey pigmentation, were noted on the lower lip and buccal mucosa. Scattered, hypopigmented macules were present on trunk and extremities.
 |  |
| Figure 1 | Figure 2 |
|---|---|
A computer tomography scan of the head showed bilateral frontal horn calcifications, which were thought to represent coronate granulomas. There was also cortical atrophy and enlargement of the left lacrimal gland. Echocardiography showed findings consistent with dilated cardiomyopathy. Colonoscopy was unremarkable.
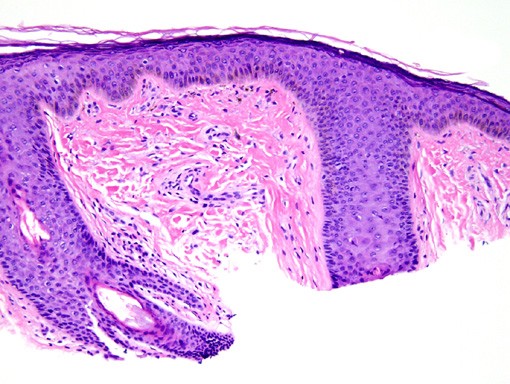
Histopathology reveals a dome-shaped lesion composed of a proliferation of blood vessels and plump, mono- and multinucleated, stellated fibroblasts.
Comment
Tuberous sclerosis (TS) is a multisystem genetic condition whose key features include multiple facial angiofibromas, hypopigmented macules, seizures, cardiac rhabdomyoma, and renal lesions. Prevalence estimates range from 1/10,000 to 1/30,000 [1, 2].
Mutations in one of two genes, TSC1 on chromosome 9q34, and TSC2 on chromosome 16p13, respectively, encoding for hamartin and tuberin, account for this disorder [3, 4]. Both genes are transmitted in an autosomal dominant fashion; however, up to two-thirds of cases may be due to spontaneous genetic mutations [5]. TSC2 mutations account for the majority of sporadic cases, and these individuals usually show a more severe phenotype that includes renal lesions and neurologic deficits [6]. The presence of germline mosaicism has also been confirmed; however, there are no standard peripheral molecular tests for detection [7].
Major features include facial angiofibromas, fibrous plaques of the forehead, hypomelanotic macules (greater than three), shagreen patches, periungual fibromas, multiple retinal nodular hamartomas, cortical tubers, subependymal nodules, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis, and renal angiomyolipoma. Minor features include multiple dental enamel pits, hamartomatous rectal polyps, bone cysts, cerebral white matter radial migration lines, gingival fibromas, nonrenal hamartoma, retinal achromic patch, confetti skin lesions, and multiple renal cysts. The presence of two major criteria or one major feature plus two minor features meet the diagnostic criteria for definite tuberous sclerosis complex [8].
Skin lesions, which are found in 70-80 percent of cases, are important features for early diagnosis and constitute four major features and one minor feature in the diagnostic criteria. Multiple facial angiofibromas, which are observed in approximately 80 percent of affected individuals, may appear within the first 2 years of life. However, angiofibromas may not be noted until adulthood, which was the case for our patient. The differential diagnosis of multiple angiofibromas should include multiple endocrine neoplasia type 1 (MEN1) and Birt-Hogg-Dube syndrome (BHDS). In addition, one should be aware that other cutaneous features in TS, such as collagenomas and mucosal fibromas, may also be observed in MEN1 and BHDS [9].
Other major diagnostic cutaneous lesions include hypopigmented macules, which are usually present at birth. Greater than three lesions must be noted to qualify as a major diagnostic feature, and configurations include thumbprint-shaped, ash leaf-shaped, and guttate or confetti-like macules. Shagreen patches represent another major diagnostic feature; they appear as leathery plaques that are frequently located on the lumbosacral region and represent a type of connective-tissue nevus. Shagreen patches typically develop around the age of 2 years and occur in approximately 50 percent of affected individuals. Periungual fibromas (Koenen tumors) are smooth, firm, skin-colored papules that are distributed around the nail folds. These periungual papules appear in late childhood and may be the sole cutaneous finding in some affected individuals. Although not included in the diagnostic criteria, cafe-au-lait macules may be found in up to 30 percent of affected individuals. Other cutaneous lesions include pedunculated, skin-colored papules (molluscum pendulum), and gingival fibromas [10].
Various ocular and neurologic manifestations occur in tuberous sclerosis and include retinal hamartomas, punched-out areas of retinal depigmentation, seizure disorders, mental retardation, learning disabilities, autism, attention-deficit disorders, and psychoses [11, 12]. Epilepsy is the most common neurologic symptom and may affect 80-90 percent of TS individuals. Supratentorial brain lesions, which are found in 90 percent of individuals, include cortical tubers, subependimal nodules, subependimal giant-cell astrocytomas, white matter linear migration lines, and transmantle cortical dysplasia. Cortical tubers represent anomalous glial proliferation and migration and are thought to have epileptogenic foci [12].
Cardiac, renal, and other organ anomalies may also occur in TS. Cardiac rhabdomyomas can be detected in 50 percent of affected infants and may spontaneously involute during the first few years of life [13]. Renal lesions are noted frequently on screening renal ultrasounds, and the majority of these lesions are angiomyolipomas. Renal cysts are less common, and renal-cell carcinomas may occasionally occur [14].
A multidisciplinary treatment approach is necessary to assess for neurodevelopmental, ophthalmologic, renal, and other organ system involvement. Neuroimaging, cutaneous and ophthalmologic examination, renal ultrasonography, echocardiography, and chest computed tomography should be done at time of diagnosis. Age-appropriate screening and monitoring for behavioral and neurodevelopmental dysfunction are also important. Periodic monitoring, which includes neuroimaging, neurodevelopmental assessment, and renal ultrasonography should be done at least every 1-3 years [15]. Many patients, including our patient, seek cosmetic treatment for the facial angiofibromas. Dermabrasion, the pulsed dye laser, and CO2 laser may be effective for angiofibromas and other fibrous lesions, which include periungual fibromas; however, recurrence rates are high [16].
References
1. Hunt A, Lindenbaum RH. Tuberous sclerosis: a new estimate of prevalence within the Oxford region. J Med Genet 1984;21:2722. Wiederholt WC, et al.. Incidence and prevalence of tuberous sclerosis in Rochester, Minnesota, 1950 though 1982. Neurology 1985;35:600
3. van Slegtnenhorst M, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997;277:805
4. The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305
5. Au KS, et al. Germline mutational analysis of the TSC2 gene in 90 tuberous sclerosis patients. Am J Hum Genet 1998;62:286
6. Jones AC, et al. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum Mol Genet 1997;6:2155
7. Rose VM, et al. Gonadal mosaicism in tuberous sclerosis. Am J Hum Genet 1997;61:A23
8. Roach ES et al. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998;13:624
9. Schaffer JV, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol 2005;53(Suppl 1):S108
10. Jozwiak S, et al. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol 1998;37:911
11. Rowley SA et al.Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol 2001;85:420
12. Curatolo P, et al. Tuberous sclerosis complex: a review of neurological aspects. Eur J Pediatr Neurol 2002; 6:15
13. Bass JL, et al. Echocardiographic incidence of cardiac rhabdomyoma in tuberous sclerosis. Am J Cardiol 1985;55:1379
14. Stillwell TJ, et al. Renal lesions in tuberous sclerosis. J Urol 1987; 138:477
15. Hyman MH, et al. National Instittues of Health consensus conference: tuberous sclerosis complex. Arch Neurol 2000;57:662
16. Boixeda P, et al. CO2, argon, and pulsed dye laser treatment of angiofibromas. J Dermatol Surg Oncol 1994;20:808
© 2007 Dermatology Online Journal