Behçet disease (incomplete) and cutaneous polyarteritis nodosa
Published Web Location
https://doi.org/10.5070/D32zc7b1bsMain Content
Behçet disease (incomplete) and cutaneous polyarteritis nodosa
Marissa Heller MD
Dermatology Online Journal 11 (4): 25
Department of Dermatology, New York University School of Medicine
Abstract
A 42-year-old man with a 2-year history of left posterior uveitis and recurrent oral aphthous ulcers presented for evaluation of recurrent, erythematous, subcutaneous, tender nodules on his lower extremities. A biopsy specimen of the nodules showed a medium-sized-vessel vasculitis with a mild and moderate inflammatory cell infiltrate in the septae and lobules of the subcutaneous adipose tissue. These changes were consistent with cutaneous polyarteritis nodosa. Behçet disease is a multisystem inflammatory disorder that includes recurrent oral aphthous ulcers, and at least two of the following features: recurrent genital ulcers, eye lesions, skin lesions, and a positive pathergy test. Behçet disease and cutaneous polyarteritis nodosa have rarely been described in conjunction, and the suggestion has been raised that cutaneous polyarteritis-nodosa-like lesions may actually be a cutaneous marker of Behçet disease.
A 42-year-old Chinese man was referred to the Dermatology Clinic at Bellevue Hospital Center in September 2004 with a 2-year history of recurrent, tender nodules on his lower extremities and of recurrent aphthous ulcers of his oral mucosa. He had recently experienced a decrease in vision in his left eye. The ophthalmologist made a diagnosis of a left posterior uveitis and referred him to the Dermatology Clinic for his other complaints. The patient denied a history of genital ulcers and had no history of gastrointestinal or neurologic complaints.
In the oral cavity on the central lower labial mucosa, right lateral labial mucosa, and left lateral tongue, there were tender, erythematous 0.5-cm ulcers with well-demarcated, edematous borders. On the lower extremities were five, erythematous, 2-3 cm, subcutaneous, tender nodules and multiple hyperpigmented patches at the sites of prior nodules. The groin was clear with no evidence of ulcers.
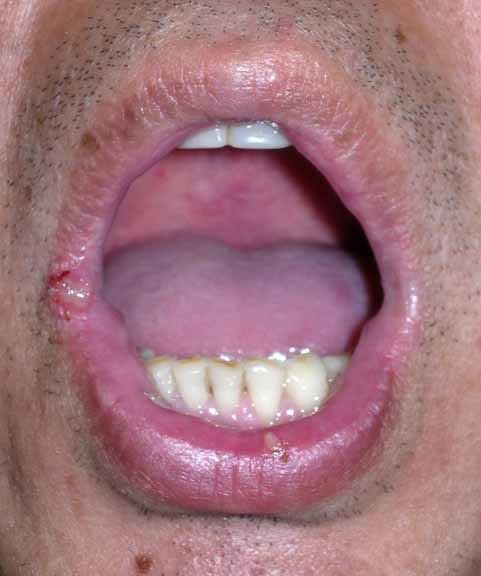
|
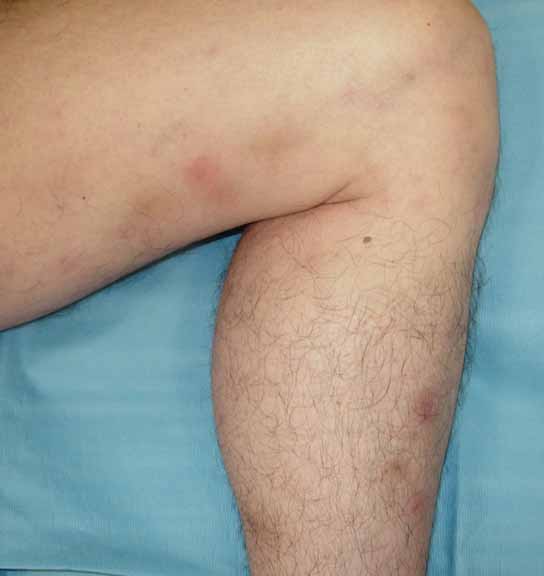
|
| Figure 1 | Figure 2 |
|---|

|
| Figure 3 |
|---|
A complete blood count, comprehensive metabolic panel, hepatic function tests, lipid profile, thyroid stimulating hormone, antinuclear antibody, erythrocyte sedimentation rate, C-reactive protein, c-antineutrophil cytoplasmic antibody (C-ANCA), p-antineutrophil cytoplasmic antibody (P-ANCA), urinalysis, hepatitis B antigen, and hepatitis-C antibody were normal or negative. A purified protein derivative (PPD) skin test was negative. His chest radiograph was normal.
Histopathology reveals within the subcutis, there is a medium-sized, thrombosed blood vessel. Neutrophils and fibrin are present with the vessel lumen and wall focally. There is an inner elastic lamina confirming that the blood vessel is an artery.
Comment
Behçet disease is a multisystem, inflammatory disorder characterized by recurrent oral aphthous ulcers and at least two of the following features: recurrent genital ulcers, eye lesions, skin lesions, and a positive pathergy test. The cutaneous lesions include erythema nodosum-like lesions, inflammatory pustules, inflammatory plaques that resemble those in Sweet syndrome, pyoderma gangrenosum-like lesions, or palpable purpuric lesions of necrotizing vasculitis. [1, 3] In addition, 10-20 percent of patients with Behçet disease develop chronic, progressive involvement of the central nervous system, and many patients also develop gastrointestinal ulcers, synovitis, and thrombophlebitis. Much of the pathologic process in Behçet disease is secondary to small-vessel vasculitis [5]. This syndrome is characterized by recurrent, self-limited attacks; however, ocular attacks can eventually lead to blindness [1, 2, 3, 4].
The syndrome is most prevalent in people from eastern Asia or the Mediterranean region, with a lower prevalence in Western countries. There is a higher incidence in men. Most often, onset is in the third or fourth decades of life. Although the cause is unknown, it is thought to be secondary to both genetic and environmental factors, through the HLA-B51 allele and a potential unidentified microbial infection, respectively. Differential diagnosis includes chronic aphthous ulcers, herpes simplex virus infection, Sweet syndrome, and inflammatory bowel disease [1].
Treatment depends on clinical manifestations and is most highly indicated with ocular lesions because 25 percent of the patients eventually become blind. Topical mydriatic agents, glucocorticoid drops, prednisone, and methotrexate are given for acute uveitis attacks, depending on the severity. Topical and systemic glucocorticoids and colchicine are used for treatment of mucocutaneous ulcerations and erythema nodosum [1, 3].
Polyarteritis nodosa (PAN) is a multisystem, necrotizing vasculitis of small-and medium-sized muscular arteries. Typically, PAN occurs in men, and the average age of onset is in the fifth decade [6]. Patients can present with muscle wasting, abdominal pain, mononeuritis multiplex, hypertension, orchitis, and congestive heart failure. Approximately 10 percent of cases of PAN are restricted to the skin. Skin lesions are typically painful, dermal or subcutaneous nodules on the lower legs and occasionally on the hands and arms. Histopathologic examination shows panarteritis that involves the small and medium-sized arteries in the dermis-subcutis junction. Clinically, the lesions can be subtle, and 20 percent of patients have concomitant peripheral neuropathy. Cutaneous PAN has been linked to infectious etiologies, such as streptococcus, parvovirus B19, human immunodeficiency virus, and hepatitis B virus infections as well as to inflammatory bowel disease and inferior vena cava thromboses. Treatment is the use of nonsteroidal anti-inflammatory agents and aspirin or high-dose glucocorticoids in more severe disease [7].
There are case reports of the rare coexistence of Behçet disease and cutaneous polyarteritis-nodosa-like lesions. These polyarteritis-nodosa-like lesions have not been included in the diagnostic criteria, but it has been suggested that they may actually be a distinct manifestation of Behçet disease [8, 9].
References
1. Sakane T, et al. Behcet's Disease. N Engl J Med 1999;341:12842. Jorizzo JL, et al. Mucocutaneous criteria for the diagnosis of Behcet's disease: an analysis of clinicopathologic data from multiple international centers. J Am Acad Dermatol 1995;32:968
3. Balabanova M, et al. A study of the cutaneous manifestations of Behcet's disease in patients from the United States. J Am Acad Dermatol 1999;41:540
4. Mangelsdorf HC, et al. Behcet's disease: report of twenty-five patients from the United States with prominent mucocutaneous involvement. J Am Acad Dermatol 1996;34:745
5. Chen K, et al. Cutaneous vasculitis in Behcet's disease: a clinical and histopathologic study of 20 patients. J Am Acad Dermatol 1997;36:689
6. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med 1997;337:1512
7. Fiorentino, DF. Cutaneous vasculitis. J Am Acad Dermatol 2003;48:311
8. Vikas A, et al. Behcet's disease with relapsing cutaneous polyarteritis-nodosa-like lesions, responsive to oral cyclosporine therapy. Dermatology Online J 2003;9:9
9. Liao YH, et al. Behcet's disease with cutaneous changes resembling polyarteritis nodosa. Br J Dermatol 1999;140:368
© 2005 Dermatology Online Journal