CHILD syndrome with thrombocytosis and congenital dislocation of hip: A case report from India
Published Web Location
https://doi.org/10.5070/D31pv2r2nhMain Content
CHILD syndrome with thrombocytosis and congenital dislocation of hip: A case report from India
Ram Chander MD1, Bincy Varghese MD1, Masarat Jabeen MD1, Taru Garg MD1, Manjula Jain MD2
Dermatology Online Journal 16 (8): 6
1. Department of Dermatology, Venereology and Leprosy2. Department of Pathology
Lady Hardinge Medical College, New Delhi, India. chanderram41@gmail.com
Abstract
Congenital Hemidysplasia with Ichthyosiform Nevus and Limb Defects (CHILD) is a very rare entity inherited as an X-linked trait. The cutaneous lesions are characteristic and usually involve the right side of the body. We report a case of CHILD syndrome in an Indian child affecting the left side with various other associations not yet described in the literature, such as thrombocytosis and congenital dislocation of the hip. The rarity of the syndrome prompted us to report this case.
Introduction
CHILD syndrome (Congenital Hemidysplasia with Ichthyosiform Nevus and Limb Defects) is a rare multisystemic disorder with about 60 cases reported in the literature. It is inherited as an X-linked trait and characterized by an inflammatory epidermal nevus showing a unique lateralization pattern, sharp midline delineation, and ptychotropism. Usually lethal in male embryos, it is recognized to be caused by a defect in cholesterol synthesis as a result of mutation of NSDHL (NADPH sterol dehydrogenase-like) gene [1, 2]. A constellation of ipsilateral extracutaneous abnormalities have been reported and include hypoplasia or aplasia of skeletal structures as well as the viscera (lung, heart, kidney, and ovary). We report a case of CHILD syndrome in an Indian neonate affecting the left side, with certain additional new clinical findings such as thrombocytosis and dislocation of the hip.
Case report
A 1-month-old girl, the second child born of a consanguineous marriage, presented with redness and scaling over the left half of the body at birth along with deformity of the left hand. There was no history suggestive of systemic involvement. The antenatal history was uneventful. The mother had no history of miscarriages and the elder male sibling was normal.
 |
| Figure 1 |
|---|
| Figure 1. Photograph showing ichthyosiform erythroderma involving the left half of the body with sharp midline demarcation |
Cutaneous examination revealed an inflammatory nevus involving the left half of the body with yellow-brown, waxy scaling over the left side of the trunk, vulva, perineum, and limbs with sharp midline demarcation. There was involvement of the rim of the pinnae and scalp with sparing of the face. The nevus diffusely involved the anterior thorax and abdomen, but showed a linear arrangement along the lines of Blaschko on the posterolateral thorax and left upper and lower limbs.
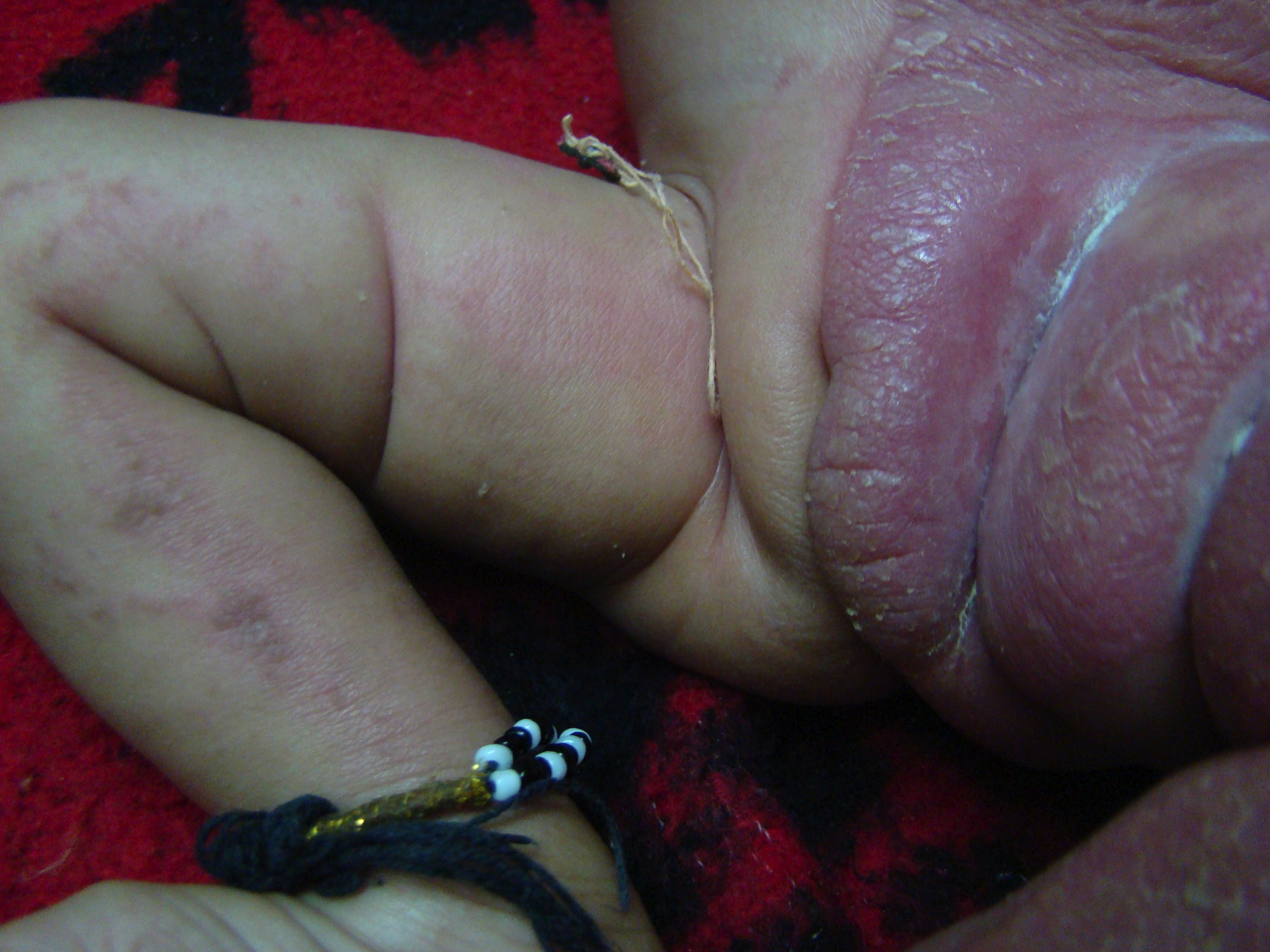
A few streaks of involvement on the contralateral side were also present. Ptychotropism in the form of involvement of the flexural creases of the axillae and groin was conspicuous. The left labium majus showed hypertrophy.
 |  |
| Figure 2 | Figure 3 |
|---|---|
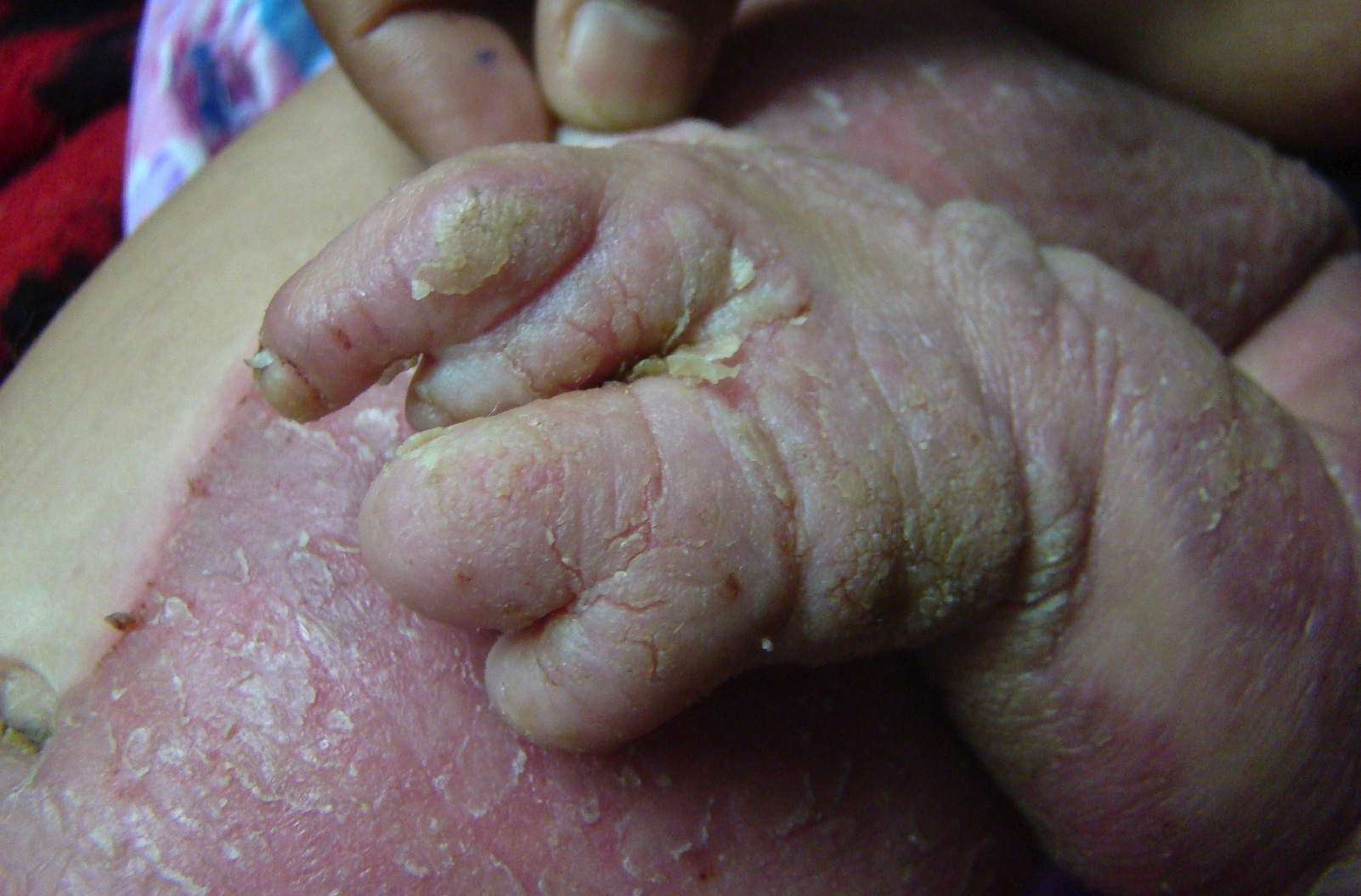
| Figure 2. Photograph demonstrating left labial hypertrophy, ptychotropism and mild streaky involvement of the contralateral
side Figure 3. Photograph illustrating lobster hand anomaly | |
Her left hand had exhibited the lobster hand anomaly. The ipsilateral foot had anonychia of the 2nd and 4th toes and dystrophy of other nails. Bilateral congenital dislocation of the hip was present. Further examination did not show any involvement of other organs such as the eyes, brain, heart, lungs, or kidneys.
Laboratory investigations revealed leukocytosis, thrombocytosis, and toxic granules within neutrophils. Chest X-ray was suggestive of subtle pneumonitis.
 |
| Figure 4 |
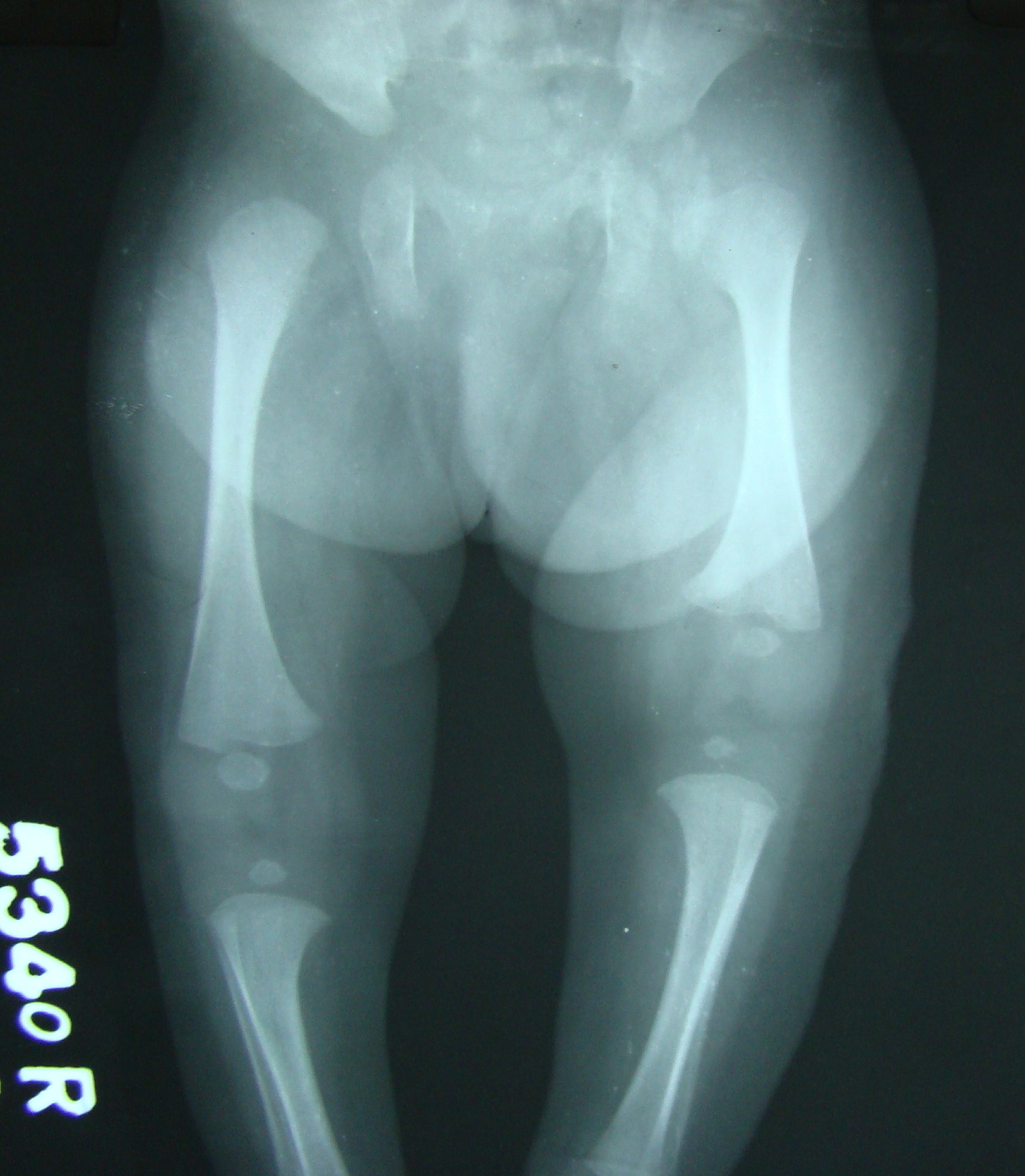
|---|
| Figure 4. X-Ray of the lower limbs showing shortening of left femur |
The infantogram showed slightly reduced length of all long bones of the left side with normal bone density. The ultrasonography of the hip joint was suggestive of left-sided congenital dislocation of the hip and proximal femoral dysplasia. Brainstem evoked response audiometry, echocardiogram, ultrasound skull, and abdomen were within normal limits.
 |
| Figure 5 |
|---|
| Figure 5. Histopathology showing orthokeratotic hyperkeratosis with parakeratosis, focal absence of granular layer, acanthosis, and mild inflammatory infiltrate in the papillary dermis |
Histological examination revealed an acanthotic epidermis with irregular elongation of the rete ridges. The thickened stratum corneum was parakeratotic with intermingled orthokeratotic strands and underlying focal absence of the granular layer. The superficial and mid-dermis exhibited a perivascular and periadnexal mononuclear inflammatory infiltrate.
A diagnosis of CHILD syndrome was made based upon the clinical and radiological findings. The patient was managed conservatively with emollients and showed mild improvement. She was discharged and advised to continue regular follow up.
Discussion
The patient we report here had left sided cutaneous lesions with ipsilateral skeletal defects along with hematological abnormalities. CHILD syndrome usually affects the right side of the body. Left sided involvement in this unique pattern of malformation is very rare, reported in only about 20 cases. The cutaneous abnormalities include unilateral erythema, scaling, and hyperkeratosis with sharp midline demarcation and ptychotropism, unilateral alopecia, and nail destruction [3]. Various commonly observed extracutaneous abnormalities in this syndrome include mild prenatal growth deficiency, limb abnormality varying from absence to hypoplasia of metacarpals, phalanges, mandible, clavicle, scapula, ribs, and vertebrae. Webbing of elbows and knees and joint contractures have also been reported associations. Other systemic abnormalities reported are ipsilateral hypoplasia of brain, cranial nerves, spinal cord, lung, thyroid, adrenal gland, ovary, and fallopian tubes. Mild mental deficiency and hearing loss were also found in a few cases. Early deaths in patients are most commonly caused by cardiovascular malformations [4, 5].
The unique features in our case include the presence of thrombocytosis and congenital dislocation of the hip, which to the best of our knowledge have not yet been described in the literature.
Until now, more than 20 different mutations including missense, nonsense, insertion, deletion, splice site, and silent mutation have been described in the NSDHL gene [6, 7, 8, 9]. Though usually lethal in male embryos, there are rare case reports of CHILD syndrome in males [10]. A rare occurrence of CHILD syndrome in 3 generations has been reported [11].
Treatment with emollients and retinoids provide little relief. However, an innovative surgical treatment using dermabrasion and skin grafting was reported to yield highly satisfactory cosmetic results in 3 patients from Germany. In 2 of these patients, the CHILD-related nevus was dermabraded, and the area was covered with split skin grafts obtained from a contralateral unaffected donor region. In a third patient, papillomatous, strawberry-like lesions on fingers and toes were excised and the defects were covered with full-thickness grafts obtained from the unaffected left gluteal area. It was suggested that the favorable outcome was related to donor dominance of the grafted skin samples that carried, in all or most cells, the mutant X chromosome in an inactivated form [12].
References
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, and Cox N, Ed. Rook’s Textbook of Dermatology, 7th ed. Oxford: Blackwell Science; 2004 : 15.1-15.1142. Bornholdt D, König A, Happle R, Leveleki L, Bittar M, Danarti R, Vahlquist A, Tilgen W, Reinhold U, Poiares Baptista A, Grosshans E, Vabres P, Niiyama S, Sasaoka K, Tanaka T, Meiss AL, Treadwell PA, Lambert D, Camacho F, Grzeschik KH. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet. 2005; 42: e17. [PubMed]
3. Happle R. Ptychotropism as a cutaneous feature of the CHILD syndrome. J Am Acad Dermatol. 1990; 23 (4): 763-766. [PubMed]
4. Shear CS, Nyhan WL, Frost P, Weinstein GO. Syndrome of unilateral ectromelia, psoriasis and central nervous system anomalies. Birth Defects. 1971; 7(8):197-203. [PubMed]
5. Christiansen JV, Overgaard Petersen H, Søgaard H. The CHILD syndrome - congenital hemidysplasia with icthyosiform erythroderma & Limb Defects. Acta Derm Venereol. 1984; 64:165-168. [PubMed]
6. Hummel M, Cunningham D, Mullett CJ, Kelley RI, Herman GE. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet. 2003; 122A (3):246-51. [PubMed]
7. Saito M, Ishiko A. A novel silent mutation in the NSDHL gene causing CHILD syndrome as a result of aberrant splicing. Br J Dermatol. 2008;159(5):1204-6. [PubMed]
8. Avgerinou GP, Asvesti AP, Katsambas AD, Nikolaou VA, Christofidou EC, Grzeschik KH, Happle R. CHILD syndrome: the NSDHL gene and its role in CHILD syndrome, a rare hereditary disorder. J Eur Acad Dermatol Venereol. 2009;24 (6):733-736. [PubMed]
9. Schmidt-Sidor B, Obersztyn E, Szymańska K, Wychowski J, Mierzewska H, Wierzba-Bobrowicz T, Stepień T. Brain and cerebellar hemidysplasia in a case with ipsilateral body dysplasia and suspicion of CHILD syndrome. Folia Neuropathol. 2008;46(3):232-7. [PubMed]
10. Happle R, Effendy I, Megahed M, Orlow SJ, Küster W. CHILD syndrome in a boy. American Journal of Medical Genetics. 1996; 62(2): 192-194. [PubMed]
11. Bittar M, Happle R, Grzeschik KH, Leveleki L, Hertl M, Bornholdt D, König A. CHILD Syndrome in 3 Generations: The importance of mild or minimal Skin Lesions. Arch Dermatol. 2006; 142:348-351. [PubMed]
12. König A, Skrzypek J, Löffler H, Oeffner F, Grzeschik KH, Happle R.Donor Dominance Cures CHILD Nevus. Dermatology. 2010. Apr 9. [Epub ahead of print] [PubMed]
© 2010 Dermatology Online Journal