Multiple eruptive keratoacanthomas, de novo
Published Web Location
https://doi.org/10.5070/D30mn783r4Main Content
Multiple eruptive keratoacanthomas, de novo
Sue Ann Wee MD
Dermatology Online Journal 10 (3): 19
From the Ronald O. Perelman Department of Dermatology, New York University
Abstract
An 89-year-old man presented with the sudden eruption of multiple, erythematous papules and nodules on his lower extremities. A skin biopsy specimen showed keratoacanthoma. After 3 months of treatment with topical imiquimod cream, the keratoacanthomas failed to resolve. We review the different clinical subtypes, histologic features, and treatment options for multiple eruptive keratoacanthomas.
Clinical synopsis
History.—The patient is an 89-year-old man with history of squamous cell carcinoma and keratoacanthomas. Five months ago, the patient developed ten erythematous, painful papules and plaques on his lower extremities. A large plaque on the left shin recently became ulcerated. He denies pruritus, bleeding, or trauma to his lower extremities. For the last 3 months, the patient has been applying topical imiquimod cream to his lower extremity plaques with some flattening and reduction in the number of lesions. The patient takes no medications, and he is otherwise healthy.
Patient denied a family history of multiple keratoacanthomas, prior keratoacanthomas in young adulthood, chemical insult, trauma, or excessive sun exposure to the lower extremities.
Physical examination.—Approximately ten erythematous plaques with scale that ranged in diameter from 0.5 cm to 1 cm were present on the right anterior lower extremity. A large 2.5 cm × 1.5-cm, ulcerated, erythematous plaque with surrounding erythema and scattered, erythematous papules with scale that ranged in size from 0.5 cm to 0.8 cm were present on the left lower extremity. Lymphadenopathy was not present.

|
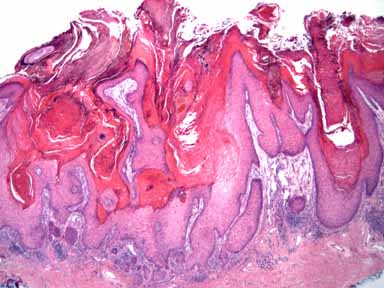
|
| Figure 1 | Figure 2 |
|---|
Laboratory data.—A complete blood count, basic metabolic panel, and liver function tests were normal.
Histopathology.—There is an exoendophytic epidermal and epithelial hyperplasia with ortho- and parakeratosis and with many keratinocytes containing abundant eosinophilic cytoplasm.
Diagnosis.—Multiple eruptive keratoacanthomas, de novo
Comment
Keratoacanthomas show rapid growth and overlapping features with squamous cell carcinomas. However, keratoacanthomas may undergo spontaneous resolution. They appear as solitary nodules with craterlike surfaces and a central keratin core and most often occur on hair-bearing, sun-exposed skin in older persons. Multiple keratoacanthomas may occur in a variety of clinical settings that include the multiple eruptive keratoacanthomas of Grybowski, the multiple, self-healing keratoacanthomas of Ferguson-Smith, and the multiple keratoacanthomas of Witten and Zak [1, 2].
It may be difficult to distinguish among the clinical subtypes. Less than 31 cases of eruptive keratoacanthomas of the Grybowski type have been reported. The average age of onset ranges from the fifth to seventh decades of life. This entity features hundreds of follicular papules with central keratotic cores on the trunk and extremities; however, larger tumors up to 5 cm in diameter, ectropion, and scleroderma-like facial changes also may occur.
The Ferguson-Smith syndrome is an autosomal dominant disorder with keratoacanthomas that appear during adolescence, spontaneously involute, and recur many years later. Keratoacanthomas of Witten and Zak display both larger and miliary-type lesions with associated ulcerative and destructive tumors. Multiple keratoacanthomas have also been reported to occur in association with the Muir-Torre syndrome [1, 3].
Histopathologic features show a symmetric tumor with a keratin-filled, cuplike crater. Generalized eruptive keratoacanthomas are histologically identical to solitary keratoacanthomas. These tumors may be indistinguishable from a well-differentiated squamous cell carcinoma. Epithelial lipping and sharp demarcation between tumor and stroma favor keratoacanthoma while ulceration and marked cellular atypia favor squamous-cell carcinoma; however, the relation between keratoacanthoma and squamous cell carcinoma remains unclear [2,4]. Although the precise etiology is unknown, dysregulation in both tumor suppressor genes (p53) and cellular oncogenes, the human papilloma virus, ultraviolet radiation, chemical carcinogenic exposure, and immune-deficiency states have been implicated [3, 4].
Systemic therapy with retinoids have been used for the multiple eruptive type of keratoacanthomas; however, recurrences and scars often occur. Solitary or early keratoacanthomas may respond to surgical excision, simple curettage with electrodesiccation, and cryotherapy with liquid nitrogen. Intralesional and topical 5-fluorouracil, intralesional glucocorticoids, and topical imiquimod also have also shown benefit. Sun exposures should be minimized as these lesions occur most often in sun-exposed skin. Despite the myriad of treatment options, there are no truly efficacious therapies [1, 3].
References
1. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Derm Surgery 2004; 30: 326.2. Schwartz RA, et al. Generalized eruptive keratoacanthoma of Gryzbowski: follow-up of the original description and 50-year retrospect. Dermatology 2002; 205: 348.
3. Consigli JE, et al. Generalized eruptive keratoacanthoma (Grzybowki variant). Br J Dermatol 2000; 142: 800.
4. LeBoit P. Can we understand keratoacanthoma? Am J Dermatopathol 2002; 24: 166.
© 2004 Dermatology Online Journal