Nephrogenic fibrosing dermopathy: First Colombian case
Published Web Location
https://doi.org/10.5070/D306h136t6Main Content
Nephrogenic fibrosing dermopathy: First Colombian case
María Soledad Aluma, Rodrigo Restrepo, Mónica Gaviria
Dermatology Online Journal 13 (3): 24
Department of Dermatology, School of Medicine, Universidad Pontificia Bolivariana, Medellín, Colombia.Abstract
A 67-year-old woman with acute renal insufficiency, who was on hemodialysis, developed progressive skin lesions consistent with thickening and hardening of the skin in both the back and extremities without contracture upon flexure. Her face was not affected. Laboratory evaluation was unremarkable and a skin biopsy showed an increase in collagen and interstitial mucin with no inflammatory infiltrate. These clinical features resemble a new recently reported disorder, nephrogenic fibrosing dermopathy. This disorder manifests itself as scleromyxedema-like skin lesions without associated paraproteinemia, occurring in the setting of renal disease. The incidence, prevalence and etiology of the disease are unknown and currently no effective treatment is available. This is the first case reported in Colombia.
Clinical synopsis
A 67-year-old patient came to the clinic because for the last 6 months she had been developing sclerotic plaques in the back and limbs. The plaques were accompanied by pruritus, pain and, a burning sensation. These lesions began to appear a month after hemodialysis treatment was initiated.
No abnormalities were detected on clinical examination. Her medical history revealed that 8 months before the present consultation she had been diagnosed with renal failure of unknown etiology. Hemodialysis (3 times weekly for 3 months) was the treatment for this condition. The patient had hypertension diagnosed 13 years before. Her oral medication included calcium, calcitriol, folic acid, enalapril, omeprazole, verapamil, and hydrochlorothiazide. She had not been exposed to gadolinium-containing contrast agents. The surgical record indicated that 1 month after being diagnosed with renal disease, the patient underwent exploratory laparotomy for an acute abdomen syndrome of uncertain etiology. No known allergies were reported.
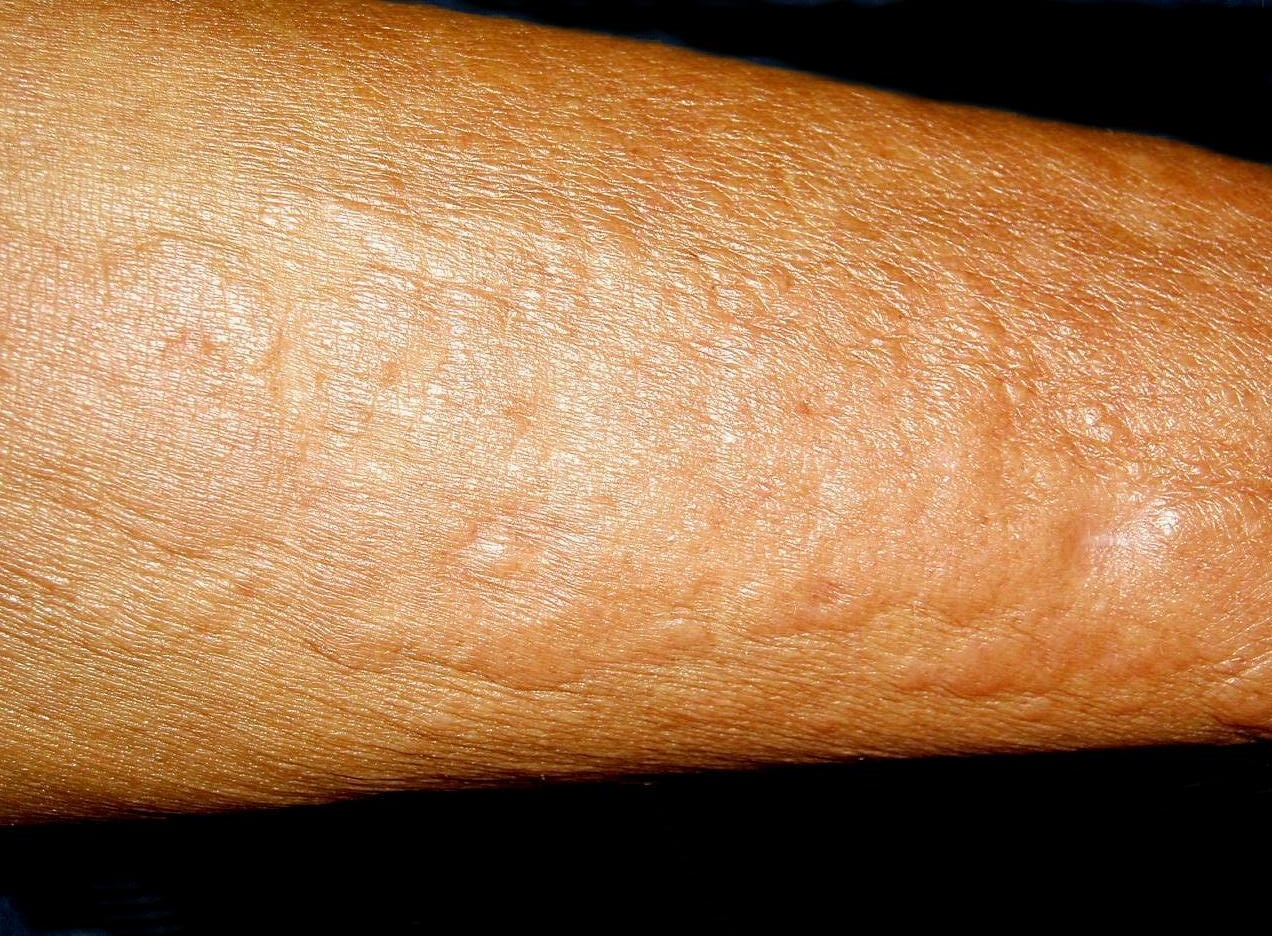
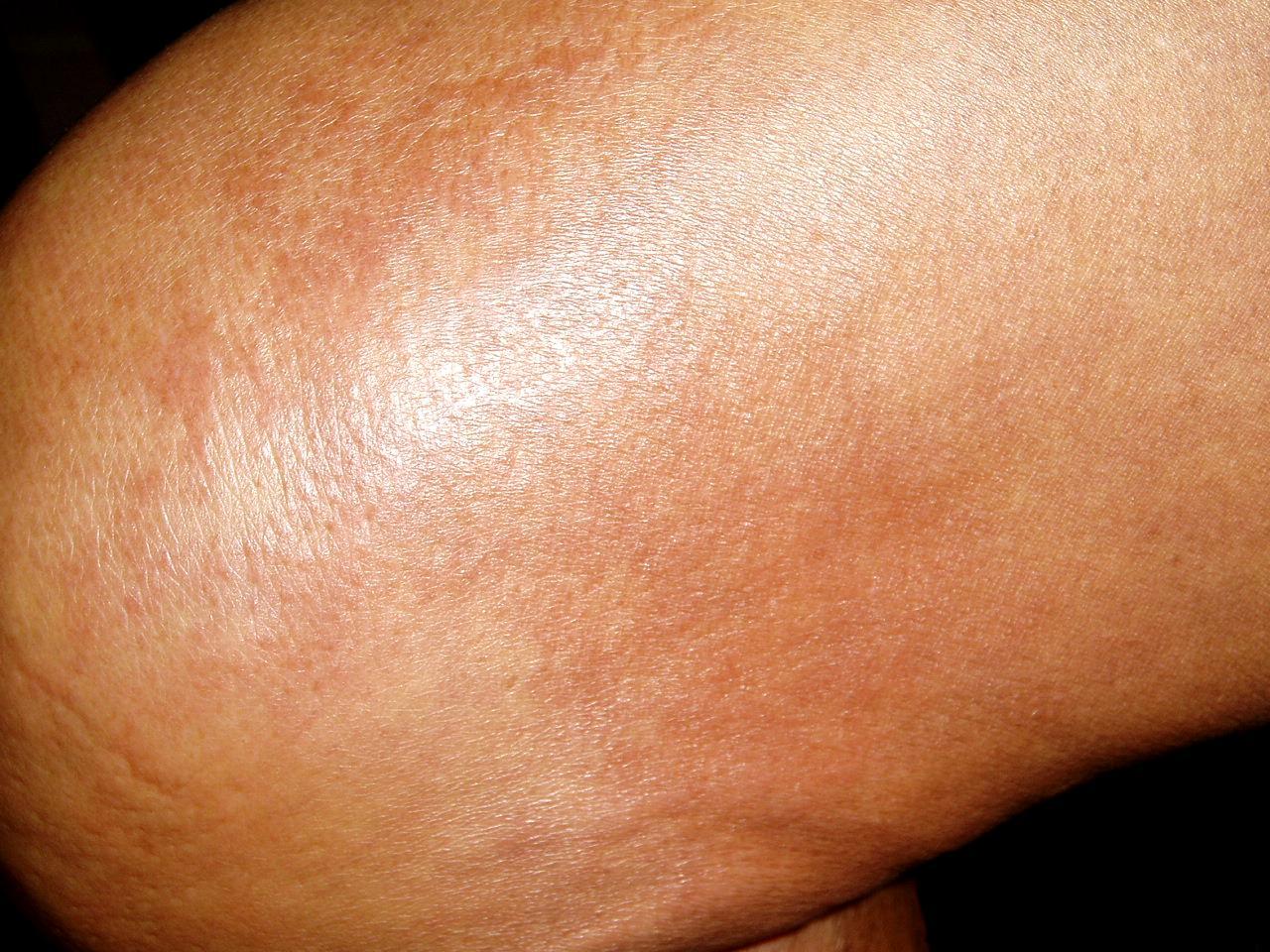
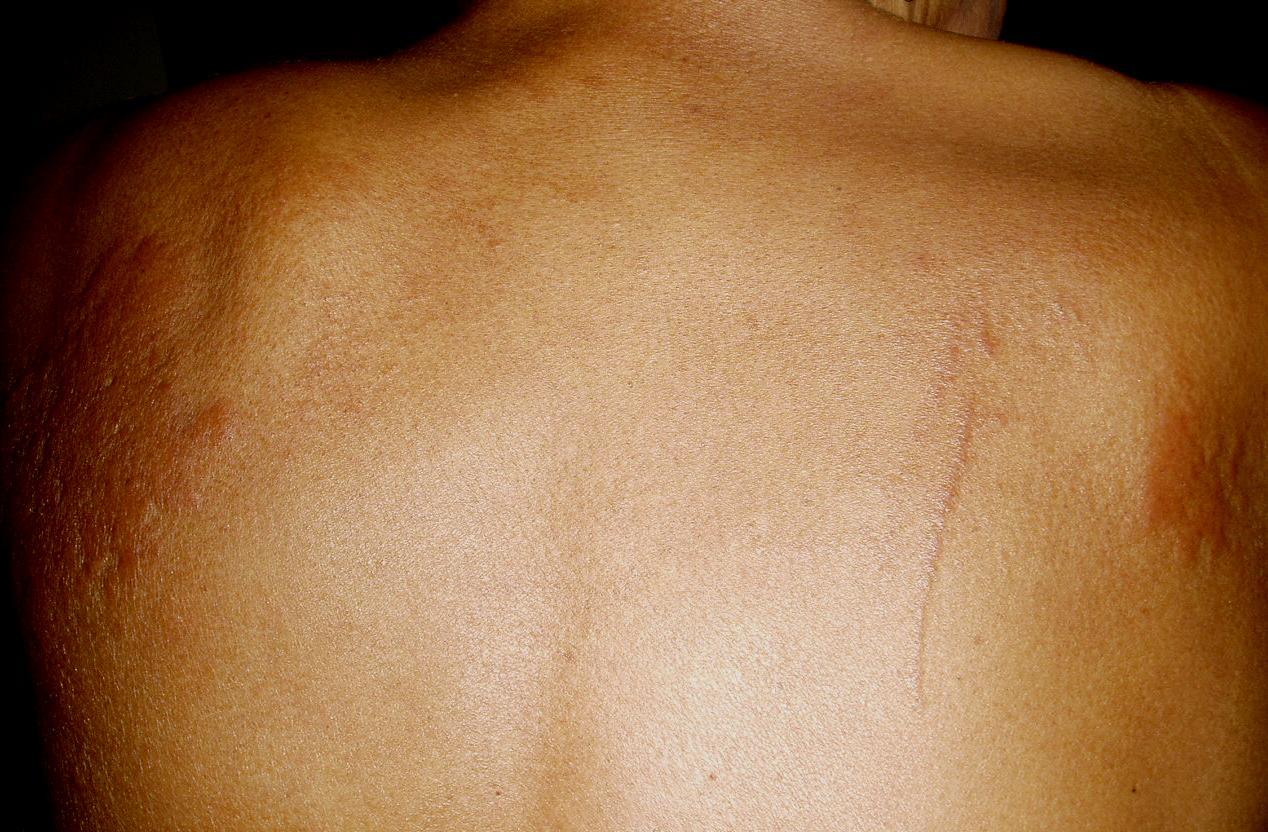
Physical examination revealed multiple sclerotic plaques with undefined borders; the lesions were slightly hyperpigmented, had a peau d'orange surface, and were localized to her forearms, thighs, legs, and back (Figs. 1 and 2)
 |  |
| Figure 1A | Figure 1B |
|---|---|
| Figure 1. Sclerotic slightly pigmented plaques with orange skin aspect located on the external face of the upper (A) and lower (B) extremities. | |
 |  |
| Figure 2A | Figure 2B |
|---|---|
| Figure 2. Sclerotic hyperpigmented plaques with the orange-skin aspect located on the thigh (A) and the back (B). | |
Her laboratory examination revealed renal impairment. She had serum creatinine levels of 2.3 mg/dl and BUN of 68 mg/dl. Blood chemistry and thyroid function tests were all within normal limits. Tests for autoimmune diseases (antinuclear antibodies, antibodies against nuclear extractable antigen, antiphospholipid antibodies, and dilute Russell viper venom time) were all negative. Protein electrophoresis was normal; an echogram of soft tissues (arm) showed an increase in the echogenicity of the subcutaneous tissues and musculature planes with no collections or calcifications.
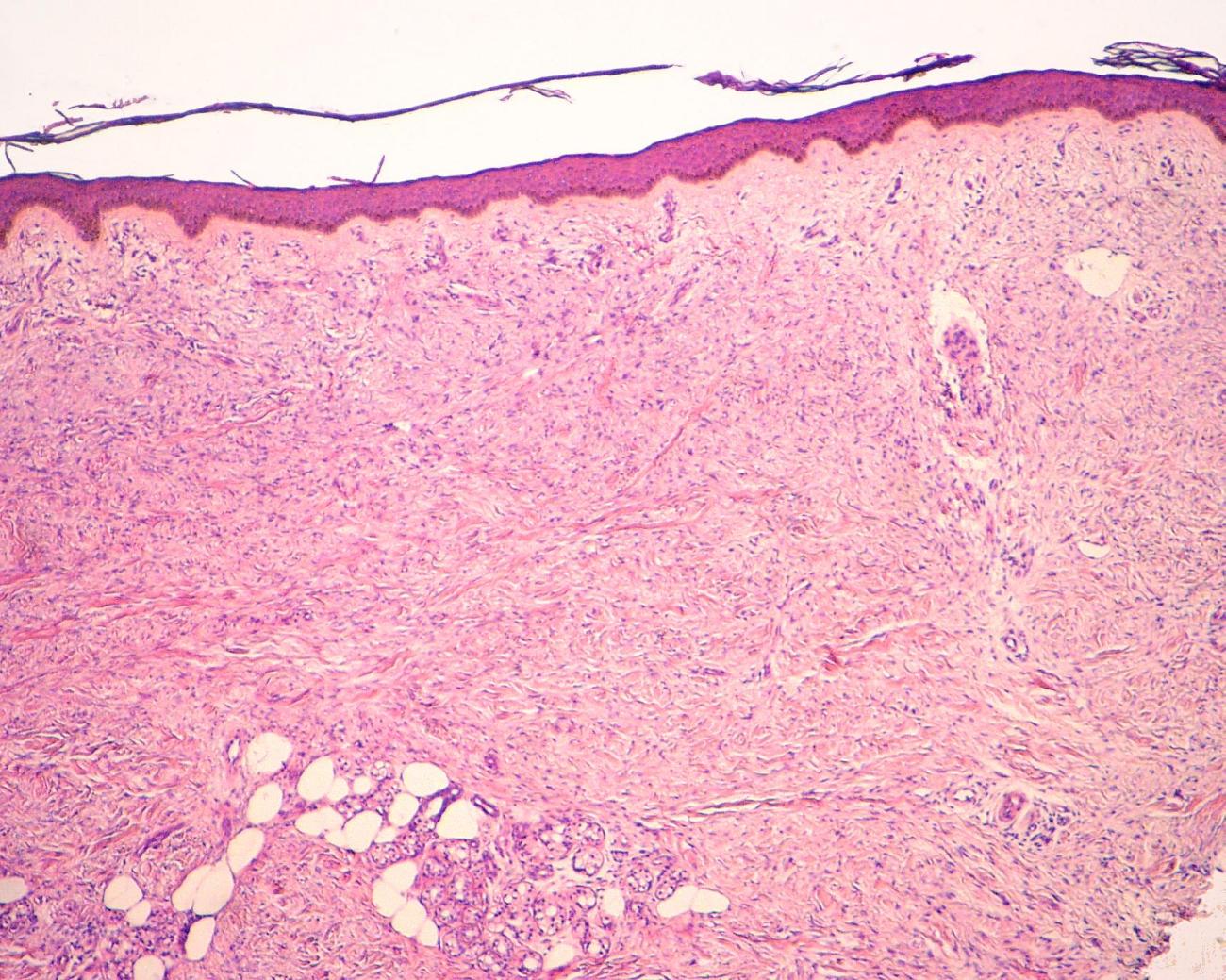
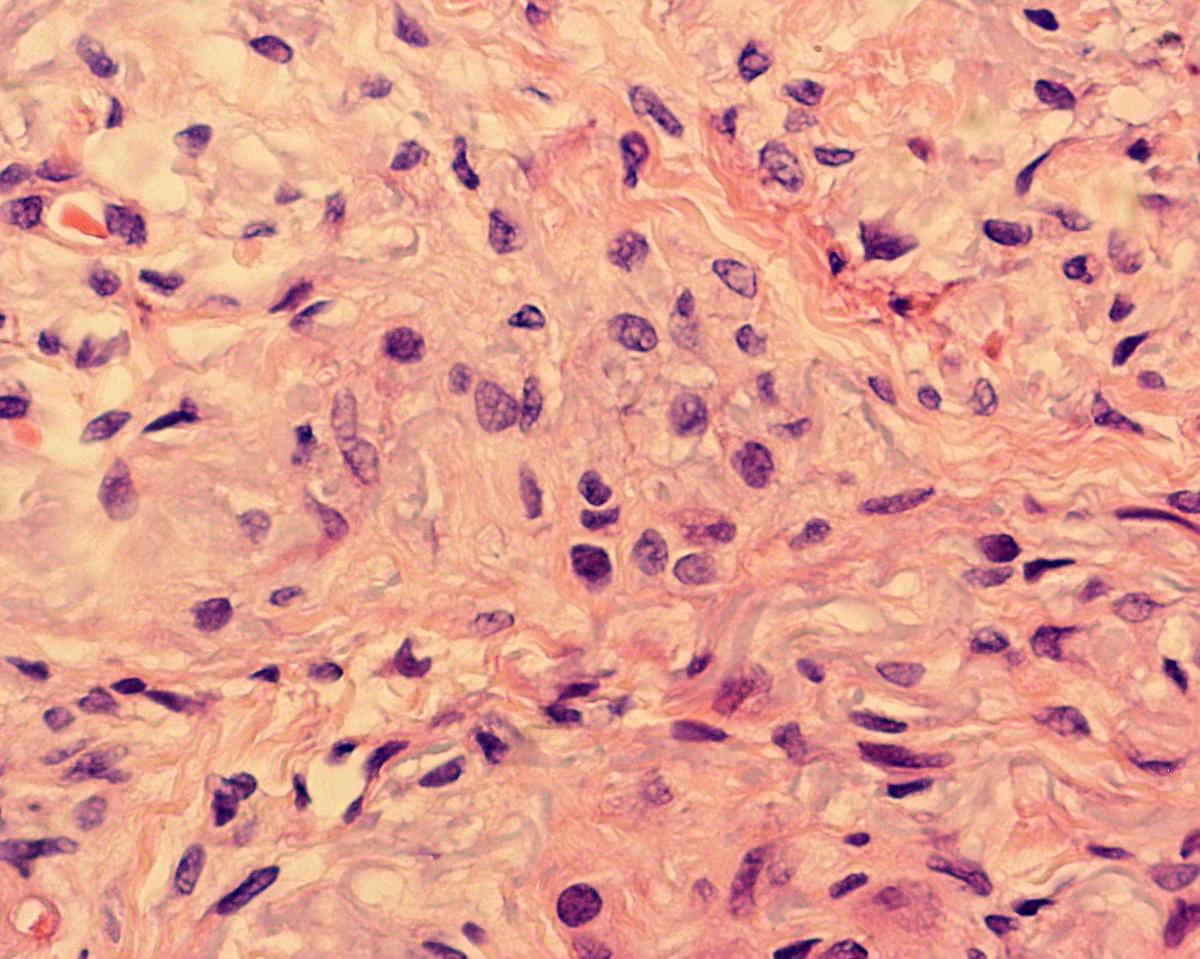
A skin biopsy was taken, fixed in buffered formalin, embedded in paraffin, cut in thin sections (3-4 microns in thickness), and stained with hematoxylin and eosin (H&E) and special immunohistochemical stains. The corresponding analysis revealed a normal epidermis and fibrous abnormalities involving the whole dermal thickness with extension to the subcutaneous tissues and replacement of the eccrine and pilosebaceous annexes (Figs. 3-5).
 |  |
| Figure 3A | Figure 3B |
|---|---|
| Figure 3. There is homogenous fibroblast proliferation and collagen deposits throughout the full thickness of the dermis (A). At higher magnification, the fuso-cellular myofibroblast component can be observed. No inflammatory infiltrate is seen (B). | |
 |  |
| Figure 4A | Figure 4B |
|---|---|
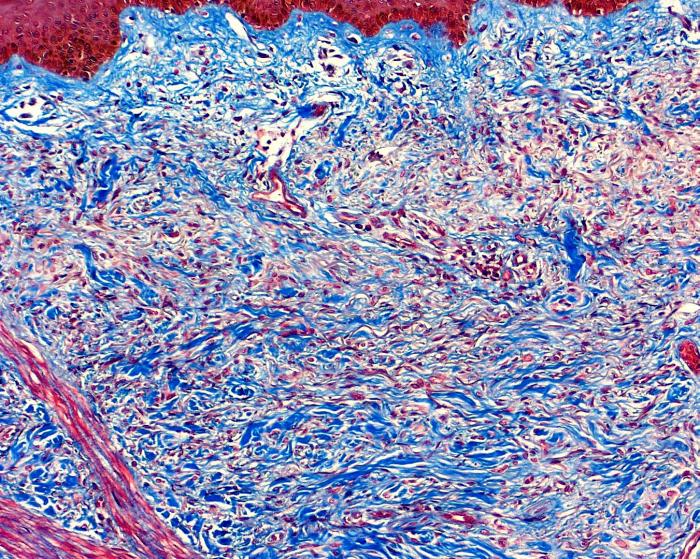
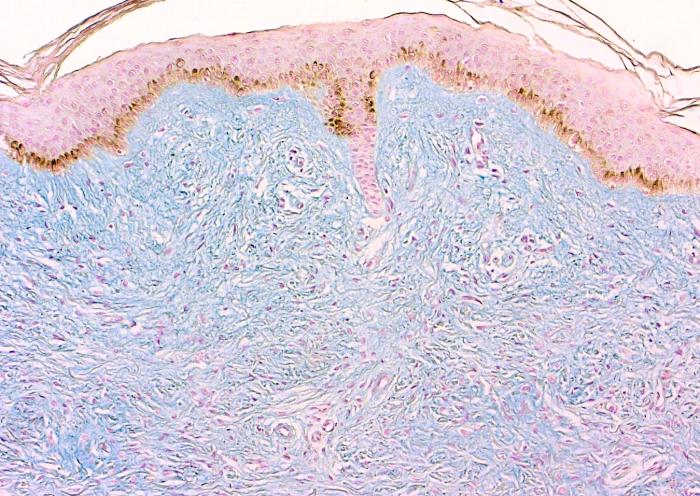
| Figure 4. Observe extensive collagen deposits with the thrichrome stain (A), and visible mucopolysaccharide with the Alcian blue stain (B). | |
The fibrosing process was localized particularly in the reticular dermis and was comprised of dense collagen and elongated cells with an oval nucleus and a myofibroblastic aspect. No inflammatory infiltrate was seen (Fig. 3). Trichrome and alcian-blue stains showed an increased connective tissue and presence of mucopolysaccharide in the dermis (Fig. 4). Stains for elastic tissue demonstrated thickening and irregular fragmentation of the elastic fibers.
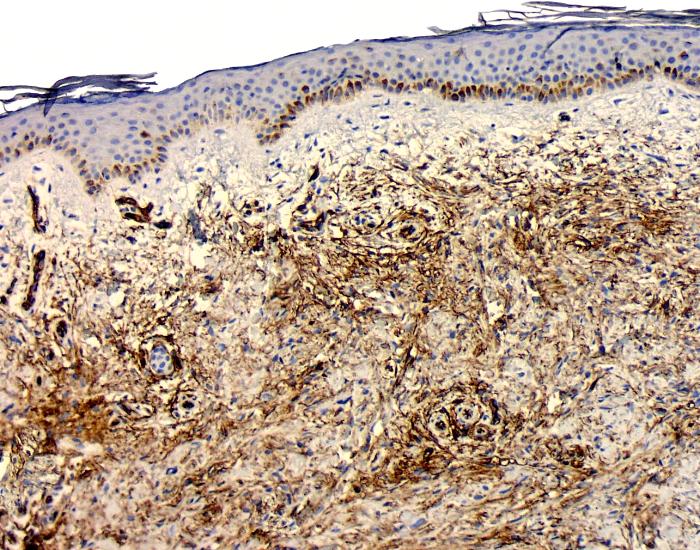
Complementary immunohistochemical studies showed strong expression of CD-34 cells throughout the lesion; these were distributed in a characteristic fibril-like pattern surrounding the collagen bundles (Fig. 5). CD68 cells were also present (Fig 6) and some of them were multinucleated.
 | 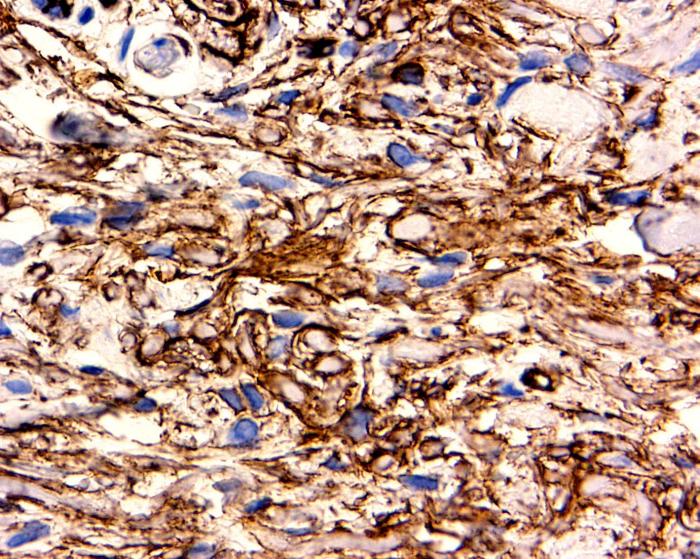 |
| Figure 5A | Figure 5B |
|---|---|
| Figures 5 Intense CD34 cells staining distributed in a fibrillary pattern, arranged around collagen fibers and blood vessels. | |
 |
| Figure 6 |
|---|
| Fig. 6. Note occasional CD68 positive cells. |
The final histopathological diagnosis was nephrogenic fibrosing dermopathy. The patient was treated with colchicine (0.5 mg every 12 hours) with a 50 percent subjective response with diminishing sclerosis, especially in the upper limbs. It should be stated that at time of this report, the patient's renal function was within normal limits.
Discussion
Nephrogenic fibrosing dermopathy (nephrogenic systemic fibrosis ), previously known as renal disease similar to scleromyxedema, scleromyxedema-type hemodialysis disease, dialysis-associated systemic fibrosis, and atypical lichen myxedematous [1] is presently considered an acquired idiotypic disease observed in patients with renal disease that clinically resembles either scleroderma or eosinophilic fasciitis, and histologically resembles scleromyxedema [2]. Despite being considered a systemic disorder, it involves preferentially the skin.
The disease was initially reported in San Francisco California in eight renal transplant patients diagnosed between May 1997 and November 2002 [3]. The corresponding biopsies were studied in the Dermatopathology Service of the University of California, where it was concluded that the abnormalities had not been reported before [3]. Shortly thereafter, Yale University created an International Center for Research on nephrogenic fibrosing dermopathy which up to June 14, 2006 has registered about 200 cases [4].
Nephrogenic fibrosing dermopathy is considered a new disease exclusive of patients with renal abnormalities independent of the etiology, severity or duration of such condition. It predominates in adults although some cases have been reported in children and elderly patients. Until now, no racial or gender predisposition has been shown. It has been identified in patients from North America, Europe, and Asia. Setting aside renal disease, other predisposing factors are hemodialysis, peritoneal dialysis, renal transplantation[5] and coagulation abnormalities. It is very common for the patient to have undergone a vascular surgical procedure or to have experienced a thrombotic episode. Recent reports (www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. and www.amershamhealth-us.com/omniscan/safety/index.html. Accessed Oct/09/2006) have correlated the development of nephrogenic fibrosing dermopathy with exposure to the MRI contrast agent, gadolinium [4]. Our patient never received this contrast agent.
Sclerotic plaques of varying size clinically characterize this disorder; nonetheless, papules and nodules have also been described. Lesions may or may not reveal hyperpigmentation but they all have the peau d'orange appearance. Limbs and trunk are the predominant localizations with the face being generally spared. The development of lesions varies from days to weeks with the patient referring pruritus, pain, and a burning sensation [6]. Incapacitating contractures and muscular weakness have been described occasionally. Other less frequent findings are palmoplantar edema with blistering and papules, as well as ocular and periorbital plaques [7].
The disease etiology is unknown. Increased numbers of fibroblasts and mucin production has been noted in all lesions. Mackay-Wiggan et al. (2003) proposed several hypotheses, one based in the observation that several patients had antiphospholipid and anticardiolipin antibodies. These autoantibodies would interact with a lipid compound or with an intrinsic substance connected with the dialysis process stimulating fibroblast and mucin production. The other hypothesis suggested that the edema presented by several patients tends to favor an increase in the number of fibroblasts [8].
Ortonne (2204) suggested that the cell responsible for this pathology was the peripheral blood fibrocyte coming from the bone marrow, a cell different from the fibroblast by its co-expression of pro-collagen I and CD34. It is thought that this new cell reaches the dermis through the circulation, and differentiates into a cell functionally and histologically similar to the normal skin fibroblast. However, the factors responsible for such differentiation are still unknown. Transforming growth factor-β has been pointed out as responsible because it is directly related with renal dysfunction [9]. Until now, no infections or toxins have been implicated. Some have proposed that peripherally deposited gadolinium might be a target for circulating fibrocytes [10].
The main abnormalities associated with the nephrogenic fibrosing dermopathy are renal and hematological. It is associated with non-specific kidney disorders, with acute renal rejection, and with sudden renal disease accompanied by edema. The hematologic alterations implicated have been abnormalities of the coagulation system, deep venous thrombosis, and recent surgery, all of which have a vascular origin [4].
Diagnosis is confirmed by means of a deep skin biopsy, an incision or punch that includes dermis, subcutaneous tissue, and fascia.
Cowper (2001) described in detail the histopathological abnormalities that characterize the disease, specifically the thickening of collagen bundles, the mucin deposits, and the proliferation of both fibroblasts and elastic fibers. During the first 20 weeks of the lesions appearance, the biopsy reveals the presence in the reticular dermis of fusiform cells that may extend to the septa and lobules of the subcutaneous fat. Such cells are distributed among the bundles of the thickened collagen and are CD34 positive. In this particular moment, the histopathologic picture may be confused with dermatofibrosarcoma protuberans or with a spindle cell melanoma [10].
A larger number of multinucleated and uninucleated positive CD68 and factor XIIIa cells are also observed. The Alcian-blue and mucicarmine stains reveal the mucin deposits. The elastic fibers, oriented parallel to the collagen bundles, appear thickened as well. Usually there are no inflammatory infiltrates. Lesions over 20 weeks old show less mucin and fewer pro-collagenous CD34 positive cells [1,2].
No other abnormalities attributable to this condition have been shown by laboratory examinations, except for renal function tests. Sporadic cases have shown positive antinuclear antibodies although the disease has not been associated with lupus; anticardiolipin and antiphospholid antibodies may also be positive in rare cases. Peripheral eosinophilia, hypercoagulability, hepatitis B and C, soft tissue calcifications and conduction abnormalities may be present. No paraproteinemia, alterations in thyroid function or positive rheumatoid factor have been demonstrated [1, 5, 11].
The two most important differential diagnoses are scleomyxedema and scleroderma. Minor resemblances may exist with morphea, eosinophilic fasciitis, eosinophilic myalgia syndrome, toxic Spanish oil syndrome, cutaneous late porphyria, fibrosing rheumatism, and β2 microglobulinemia amyloidosis [6, 12, 13]. All the above diseases are characterized by dermal fibrosis, but with the exception of scleroderma, none is associated regularly with renal disease [7]. Scleromyxedema affects particularly the face, arms, and hands but not the legs or the trunk. This disease is considered as a variant of lichen myxedematosus and frequently has systemic manifestations. Usually, it is accompanied by monoclonal paraprotein. Histologically it closely resembles nephrogenic fibrosing dermopathy, with the exception of the presence of an important inflammatory infiltrate of the lymphocyte-plasmocytic type and of a notorious quantity of interstitial mucin. Scleroderma is characterized by both systemic and skin involvement, frequently associated with Raynaud phenomenon and with circulating auto-antibodies; histologically, collagen is thickened with loss of the spaces between the corresponding fibers. The inflammatory infiltrate varies according to the stage of the disease, but there is no proliferation of the CD34 positive cells, so characteristic of the nephrogenic fibrosing dermopathy, nor is dermal mucin increased. Eosinophilic fasciitis affects especially the distal aspects of the extremities; it is associated with systemic symptoms and 75 percent of the patients present polyclonal hypergammaglobulinemia accompanied by peripheral eosinophilia. Histological findings differ greatly from those of the fibrosing dermopathy and consist of fascia infiltrates made of mononuclear and eosinophilic cells, resulting in edema, thickening and sclerosis. No mucin deposits are observed and there is no renal involvement. The esosinophilic-myalgia syndrome is related to ingestion of contaminated L-tryptophan. Skin manifestations are variable and include damage to the extremities and occasionally, to the trunk and face. Peripheral eosinophilia and incapacitating myalgias are present. Histologically, findings are similar to those of scleroderma with inflammatory infiltrates that include lymphocytes, plasma cells, macrophages, and eosinophils. The toxic Spanish-oil syndrome is related to non-consumable oil contaminated with aromatic amines. It is a multisystemic disorder that histologically shows a lympho-histiocytic infiltrate located both in the superficial and deep layers associated to vascular changes that may go from endothelial edema to small-artery vasculitis. Occasionally, large quantities of mucin deposits can be observed.
Concerning therapy, numerous treatments have been used for nephrogenic fibrosing dermopathy without avail as no compound appears to significantly help. Reports about favorable response are anecdotal. Renal function improvement retards the disease course or may reverse it. Those treatments that have shown better results are prednisone (1 mg/kg)[1], calcipotriol[4], IV immunoglobulin[14], photopharesis [15], photodynamic therapy [16], plasmapharesis, and renal transplant [4]. Other studies have described little improvement with thalidomide, PUVA [17], intralesional methrotrexate, triamcinolone, intralesional α-interferon, pentoxifylline, and cyclophosphamide. Physical therapy has been used in patients with contractures [1, 4].
It is prudent to exclude the use of gadolinium in patients with renal disease (www.fda.gov/cder/drug/advisory/gadolinium_agents.htm).
Prognosis is unknown; the disease is considered a progressive disorder, albeit gradual improvement and spontaneous recovery upon renal disease control have been described. Recently, however, cases with systemic involvement of organs such as diaphragm, esophagus, the psoas muscle, renal and lung vasculature have been shown to have a bad prognosis [18].
In summary, we have presented the case of a woman with renal disease treated with hemodialysis who presented nephrogenic fibrosing dermopathy, this being the first case described in Colombia.
References
1. Sheinfeld N. and Cowper S. Nephrogenic Fibrosing Dermopathy. Available at http://www.emedicine.com/derm/topic934.htm. Accessed Oct/09/2006.2. Cowper SE, Su L, Robin H, Bhawan J, LeBoit PE. Nephrogenic fibrosing dermopathy. Am J Dermatophatol 2001; 23:383-393.
3. Cowper SE. Fibrosing skin conditions among patients with renal diseases - US and Europe 1997 – 2002. MMWR/Morb Mortsl Wkly Rep 2002; 51:25–26.
4. Cowper SE. Nephrogenic Fibrosing Dermopathy [NFD/NSF Website]. 2001-2006. Available at http://www.icnfdr.org. Accessed Oct/09/2006
5. Swartz RD, Crofford LJ, Phan SH, Ike RW, Su LD. Nephrogenic fibrosing dermopathy: A novel cutaneous fibrosing disorder in patients with renal failure. Am J Med2003; 114 :563–572.
6. Streams BN, Liu V, Liegeois N, Moschella SM. Clinical and pathological features of nephrogenic fibrosing dermopathy. J Am Acad Dermatol 2003; 48:42-47.
7. Leboit PE. What Nephrogenic fibrosing dermopathy might be. Arch Dermatol 2003; 139:928–30.
8. Mackay – Wiggan JM, Cohen DJ, Hardy MA, Knobler EH, Grossman ME. Nephrogenic fibrosing dermopathy (scleromyxedema – lyke illness of renal disease). J Am Acad Dermatol 2003; 48:55–60.
9. Ortonne N, Lipsker D, Chantrel F, Boehm N, Grossman E et al. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J of Dermatol 2004; 150:105–102.
10. Cowper SE, Bucala R, Leboit PE. Nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis-setting the record straight. Semin Arthritis Rheum. 2006; 35(4):208-10.
11. Edsall LC, English JC 3rd, Teague MW, Patterson JW. Calciphylaxis and metastasic calcification associated with nephrogenic fibrosing dermopathy. J Cutan Pathol 2004; 31:247–253.
12. Moschella SL, Kay J, Mackool BT, Liu V. Case 35-2004: A 68 year old man with end-stage renal disease and thickening of the skin. N Engl J Med 2004; 351:2219–2217.
13. Markus JS, James AJ, Nuñez-Gussman JK, Sheehan AM, Fegan L. et al. Nephrogenic fibrosing dermopathy. J Am Acad Dermatol 2005; 52:166-7.
14. Chung HJ, Chung KY. Nephrogenic fibrosing dermopathy: response to high–dose intravenous immunoglobulin. Br J Dermatol 2004; 150: 596–597.
15. Lauchli S, Zortea-Caflisch C, Nestle FO, Burg G, Kempf W. et al. Nephrogenic fibrosing dermopathy treated with extracorporeal photophoresis. Dermatology 2004; 208: 278–280.
16. Schmook T, Budde K, Ulrich C, Neumayer HH, Fritsche L. et al. Successful treatment of nephrogenic fibrosing dermopathy in a kidney transplant recipient with photodynamic therapy. Nephrol Dial Transplant 2005; 20:220-222.
17. Kafi R, Fisher GJ, Quan T, Shao Y, Wang R, et al. UV-A phototherapy improves nephrogenic fibrosing dermopathy. Arch. Dermatol 2004; 140:1322–4.
18. Ting WW, Stone MS, Madison KC, Kurt K. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol 2003; 139:903–906.
© 2007 Dermatology Online Journal