Case of mistaken identity: Bullous congenital ichthyosiform erythroderma mistaken as epidermolysis bullosa simplex
Published Web Location
https://doi.org/10.5070/D301h2c4k6Main Content
Case of mistaken identity: Bullous congenital ichthyosiform erythroderma mistaken as epidermolysis bullosa simplex
Ali Alikhan MD1, Dina Farshidi 2, Tor Shwayder MD3
Dermatology Online Journal 15 (9): 3
1. University of California Davis, School of Medicine, Sacramento, California2. University of California Los Angeles, David Geffen School of Medicine, Los Angeles, California
3. Henry Ford Hospital, Department of Dermatology, Detroit, Michigan. TShwayd1@hfhs.org
Abstract
Distinguishing clinically similar dermatologic disorders can be challenging and the differential diagnosis of a blistering eruption in the newborn period can be extensive. Several genodermatosis, such as bullous congenital ichthyosiform erythroderma (BCIE) and epidermolysis bullosa simplex (EBS), autoimmune bullous disorders, infectious diseases, sucking blisters, and bullous mastocytosis must be considered. We present a case of BCIE misdiagnosed as EBS and review characteristic clinical and histopathological features of each disorder as well as the basic approach to treatment.
Introduction
Frequently, similar-appearing but etiologically different dermatologic disorders are mistaken for each other. Thus, understanding what makes each disease unique can help the clinician differentiate between phenotypically similar disorders. This is especially true for epidermolysis bullosa simplex (EBS) and bullous congenital ichthyosiform erythroderma (BCIE).
Epidermolysis bullosa (EB) is a group of inherited blistering disorders with an estimated prevalence between one and three per 100,000 children; EBS is a subtype of EB characterized by intraepidermal blistering secondary to keratin gene mutations [1]. The three most common EBS subtypes, generalized (Koebner), localized (Weber-Cockayne), and herpetiform (Dowling-Meara), are each inherited in an autosomal dominant fashion [2].
BCIE, also known as epidermolytic hyperkeratosis, occurs in 1 out of every 100,000-300,000 live births and appears in the neonatal period with blistering, scaling, and redness. It progresses to generalized hyperkeratotic scaling in infancy and adulthood [3].
Our case of BCIE demonstrates the difficulty of differentiating blistering disorders in the newborn period when a case of BCIE can easily mimic other blistering disorders such as EBS. As with many other genetic disorders, patients with BCIE present with different clinical and histopathologic findings as newborns than as children and/or adults. Subtle changes can be overlooked; during the natural progression of the disorder valuable information can be lost if not specifically searched for by an experienced clinician who can piece together clinical and histopathologic findings. Genetictesting may be requiredto make the correct diagnosis early in the patient's life.
Case report
A 4-year-old boy presenting with generalized 'rash' since birth was misdiagnosed as having epidermolysis bullosa simplex (EBS). The initial biopsy taken several years earlier by a non-dermatologist was signed out as EBS. We do not have access to the original pathology slides and were only able to gather information from the patient's records from an outside clinic. These reports stated that the initial biopsy showed an "intraepidermal blister within the basal epidermal layer, along with clumping of the tonofilaments." This is a second hand report of the biopsy results and we are left to assume that clumping of the tonofilaments was determined by electron microscopy. A second reading of the biopsy by an EBS expert noted no definite cleavage planes and normal type VII collagen. These findings excluded generalized recessive dystrophic EB and junctional EB, but left EBS in the differential.
Although his clinical appearance did not fit EBS, the patient was seen and misdiagnosed by various non-dermatologists in the US. Working diagnoses ranged from EBS to BCIE to severe seborrheic dermatitis, highlighting the importance of clinicopathologic correlation by a trained specialist in difficult-to-diagnose cases such as this. His mother believed that flaring of the 'rash' was secondary to specific foods in his diet such as chicken, dairy products, eggs, and milk. However, the flares did not improve with elimination of these food items from his diet. Furthermore, dietary restrictions may have resulted in failure to thrive and a subsequent intervention by social services. His developmental milestones were normal.
In addition, the patient had been hospitalized five times in the last eight months for methacillin-resistant Staph aureus (MRSA) infections. He had also lost six pounds over the last year, and weighed less than he did at his third birthday. His mother had tried various treatment options including topical application of Aquaphor™ ointment, intensive moisturizing with Cetaphil™ lotion and oral acitretin, without significant improvement. There was no family history of dermatologic disease.
 |  |
| Figure 1 | Figure 2 |
|---|---|
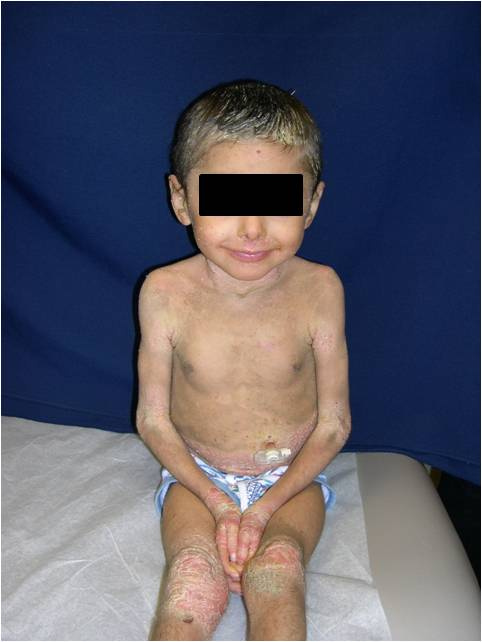
| Figure 1. Our patient with BCIE at 4 years. Note, hyperpigmented, dirty-appearing scale with increased verrucous-warty linearity
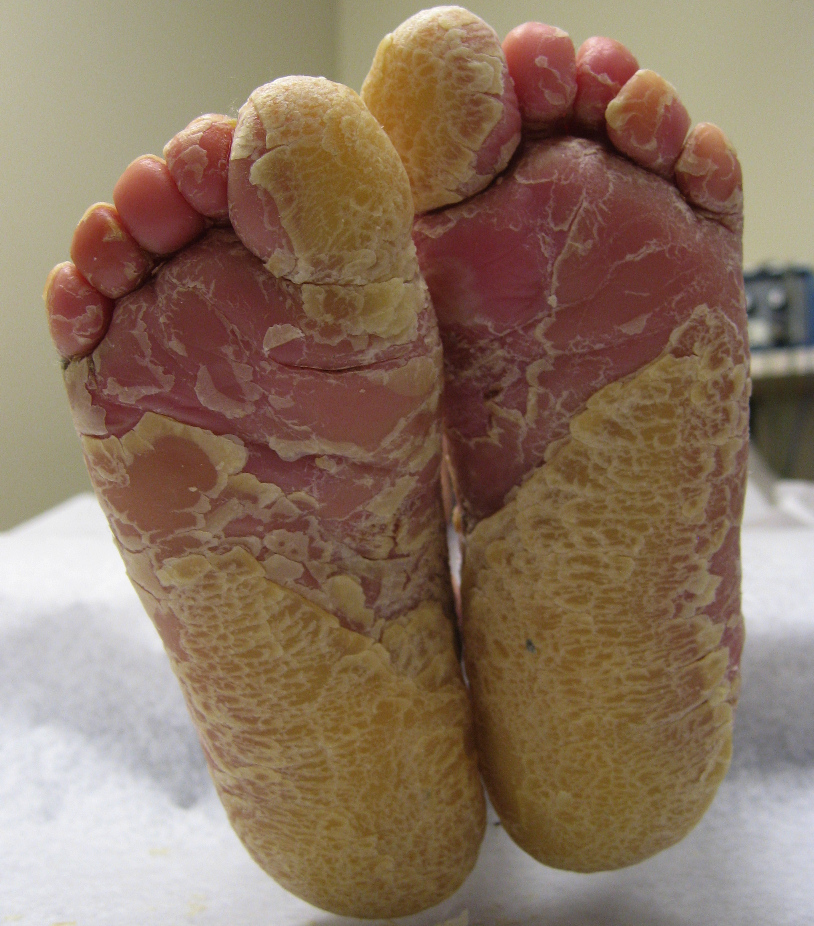
especially in the flexural surfaces of the axillae, popliteal, and antecubital fossae. Figure 2. Our patient with BCIE at 4 years. Note, thick yellow plantar keratoderma. | |
On presentation, at 4 years of age, physical examination revealed hyperpigmented, dirty-appearing scale with increased verrucous-warty linear plaques, especially in the flexural surfaces of the axillae, popliteal, and antecubital fossae (Fig. 1). There was also thick yellow scale and palmoplantar keratoderma involving the hands and feet bilaterally (Fig. 2), and thick yellow-matted scale encompassing the scalp. Some degree of scale was present throughout all body surfaces, with approximately 95 percent involvement. Fissuring was present on the hands, feet, and angles of the mouth. No blisters were noted and there was no involvement of the ocular mucosa. A generalized foul odor emanated from the child.
At this time, on the basis of clinical presentation alone, it was evident that EBS was not the correct diagnosis. The total body warty, porcupine quill-like hyperkeratosis, palmar hyperkeratosis, and thick scalp scale were characteristic of BCIE. A repeat biopsy showed epidermal acanthosis, papillomatosis and hyperkeratosis. There was striking clumping and dissolution of the granular cell layer, both consistent with BCIE.
For treatment of his scalp, we prescribed olive oil to be generously applied under shower cap occlusion for approximately 2-3 hours prior to washing with T/Gel shampoo. To his body, the patient was instructed to apply Vaseline™, Aquaphor™, and Crisco™ after bathing at a recommended twice daily frequency to improve moisturization. Furthermore, a small quantity of liquid laundry bleach was added to the bath water (¼ to ½ cup per foot of water on side of tub) to kill and eliminate cutaneous bacteria responsible for the associated foul odor. Oral anti-staphylococcal antibiotics were also used for purulent lesions. Oral retinoids, including isotretinoin and etretinate, are another treatment option that is relatively fast acting and effective for this condition; in many cases retinoids would provide benefits that would arguably be greater than any of the associated potential side effects (e.g. skeletal abnormalities such as cortical hyperostoses, diminished bone density and premature epiphyseal closure) [4, 5, 6, 7]. However, given that our patient was young, that his condition required long-term treatment and that he also suffered from baseline growth retardation, which had necessitated a percutaneous endoscopic gastrostomy (PEG) tube placement and overnight feeds, it was decided that we would first start treatment with topical therapy and oral antibiotics. Retinoids could be considered at a later time if the patient's dermatologic condition failed to improve. Thus far, oral antibiotics along with the bleach baths have helped control and reduce skin infections and have consequently also helped the associated foul odor. Furthermore, extensive use of emollients has helped soften and remove excess scale from his body and scalp.
Discussion
Hereditary EB comprises a spectrum of rare genodermatoses in which deformed epidermal and/or dermal proteins lead to blister formation following trauma. Presentations range from mild to disabling, and differ in inheritance, phenotype, and associated findings [2]. EB can be subdivided into categories based on the ultrastructural level at which blister formation occurs; this is best observed on transmission electron microscopy (TEM). These groups include simplex, junctional, dystrophic, and hemidesmosomal EB [8]. In each group, there exist numerous subtypes, each with unique clinical appearance, histological findings, and genetic derangements [2].
 |  |
| Figure 3 | Figure 4 |
|---|---|
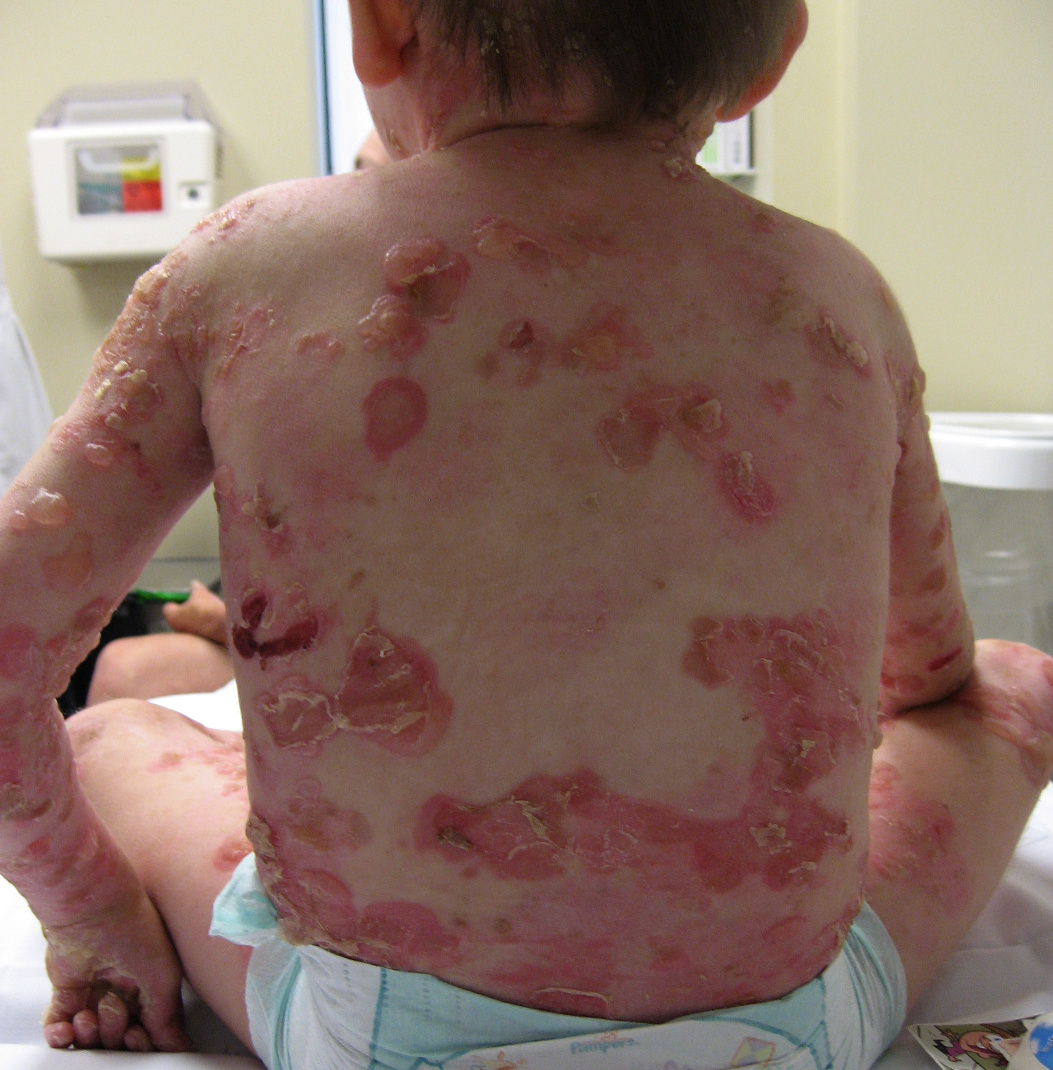
| Figure 3. An EBS patient at birth. Note, vesicles and bullae over upper and lower extremities. Figure 4. An EBS patient at 37 months. Note, vesicles and bullae over back and upper extremities. | |
Within EBS, the EB subgroup with tissue separation within the basal keratinocytes, there are three major subtypes. The generalized subtype of EBS (Koebner) is characterized by generalized blistering without clustering. Vesicles and bullae appear predominantly on the palms and soles, and in areas of trauma at birth (Fig. 3) [8]. These initial lesions usually improve or resolve but later recur when the child becomes more active (Fig. 4) [2, 8, 9]. The disease is also reportedly worse during the warmer summer months. This subtype of EBS is typically milder than other forms of EBS and not associated with major disability.
The localized subtype of EBS (Weber-Cockayne) typically presents with bulla almost exclusively on the hands and feet at sites of trauma, friction and increased pressure. Most cases are diagnosed in early childhood as the child begins to crawl and walk, although mild cases may go undiagnosed until adulthood [2, 8]. Oral soft tissue erosions have also been reported in approximately 25 percent of cases [2]. Patients may also complain of pain and blistering where shoes rub against the feet, worsening of blistering after prolonged walking or marching, and mild hyperkeratosis of plantar surfaces of the feet from chronic blistering [9].
Finally, the herpetiform variant of EBS (Dowling-Meara) manifests at birth with widespread blistering that over a period of months takes on a more clustered, arciform or "herpetiform" pattern and may heal with milia or mild atrophic scarring [10]. Dowling-Meara can also be associated with involvement of the oral mucosa, dystrophic or absent nails, and a progressive diffuse palmoplantar keratoderma [2, 11]. When the oral cavity is involved, difficulty with chewing and swallowing may follow and lead to a secondary state of anemia, malnutrition and growth retardation [2]. Clinically, Dowling-Meara is the most severe subtype of EBS and extensive skin involvement can also be followed by secondary skin infections, sepsis and respiratory failure, rarely culminating in death [10, 12].
Our patient was initially thought to have generalized EBS, which typically presents with blistering at birth or during early infancy [2]. It is dominantly inherited, and as described above, blisters occur following trauma and thus favor high impact areas such as the feet, hands, elbows, and knees [2, 9]. Furthermore, palmoplantar hyperkeratosis, which can be a late feature of EBS with onset in late childhood, is also a feature of BCIE. Nails, teeth, and oral mucosa are generally spared.
 |
| Figure 5 |
|---|
| Figure 5. Punch biopsy of EBS lesion demonstrates intact stratum corneum and upper epidermis, with vesicle formation in the lower epidermis at the basal layer caused by degeneration of individual epidermal cells. |
Most EBS patients have mutations in the genes coding for K5 and K14 keratins, with the degree of clinical severity depending on the region of the gene affected [2]. Keratin 5 and keratin 14 form heterodimers that support the structure of the basal cells in the epidermis. Thus mutations in these two polypeptides can disrupt the stability of the keratin filament network that allows the keratinocyte to withstand mechanical forces, resulting in cytolysis of basal keratinocytes and separation or clefting of the basal cell layer [13]. Light microscopy may show skin separation at the basal cell level, and may also demonstrate variable intermediate filament clumping (Fig. 5) [14]. Often electron microscopy is used in conjunction with or in place of light microscopy to better delineate the exact location of the intraepidermal blister. With electron microscopy, keratin filament aggregates also known as tonofilaments may also be seen within basal cells and are characteristic for Dowling-Meara EBS [14]. Immunoflourescence antigenic mapping using monoclonal antibodies specific for K5 and K14 can also be used to diagnose EBS and is an important method of diagnosis given its rapid turnaround time and high sensitivity and specificity [15]. Most gene mutations associated with EBS are dominantly inherited, although recessive transmission has been reported [8]. Mutational analysis is currently available and a valuable tool in prenatal diagnosis [2, 16]. Genetic testing for the two genes associated with EBS, KRT5 and KRT14, are commercially available in the US (www.genedex.com) [17].
There is no definitive cure for EBS and management is based mainly on anecdotal and case reports. Currently, treatment regimes are tailored to the meet the needs of the individual patient, taking into consideration severity and extent of involvement. Most treatment plans consist of supportive care for the skin, appropriate management of other organ systems that may be involved, and systemic therapies such as antibiotics for complications such as infections [2]. Skin care for blistering and erosions may include saline compresses, topical antibiotics, and topical steroids, with emphasis on gentle bathing and cleansing techniques followed by application of protective emollients and non-adherent dressings. Unfortunately, few randomized controlled trials (RCTs) looking at treatments for inherited forms of EB exist. This short list includes two studies on tetracyclines and a study each on topical aluminum chloride hexahydrate solution 20 percent, topical bufexamac cream 5 percent, and phenytoin [18]. A recent review of these RCTs found no clear benefit to any of the treatment interventions studied [18].
 |
| Figure 6 |
|---|
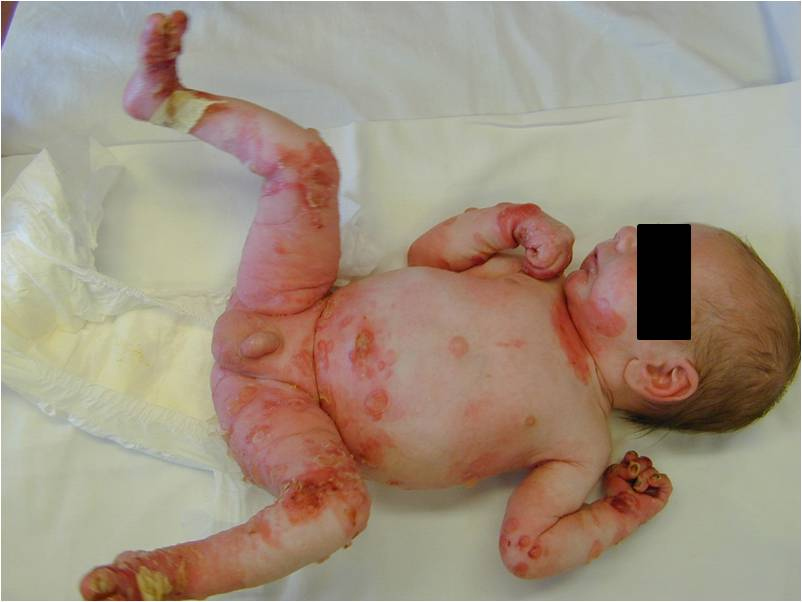
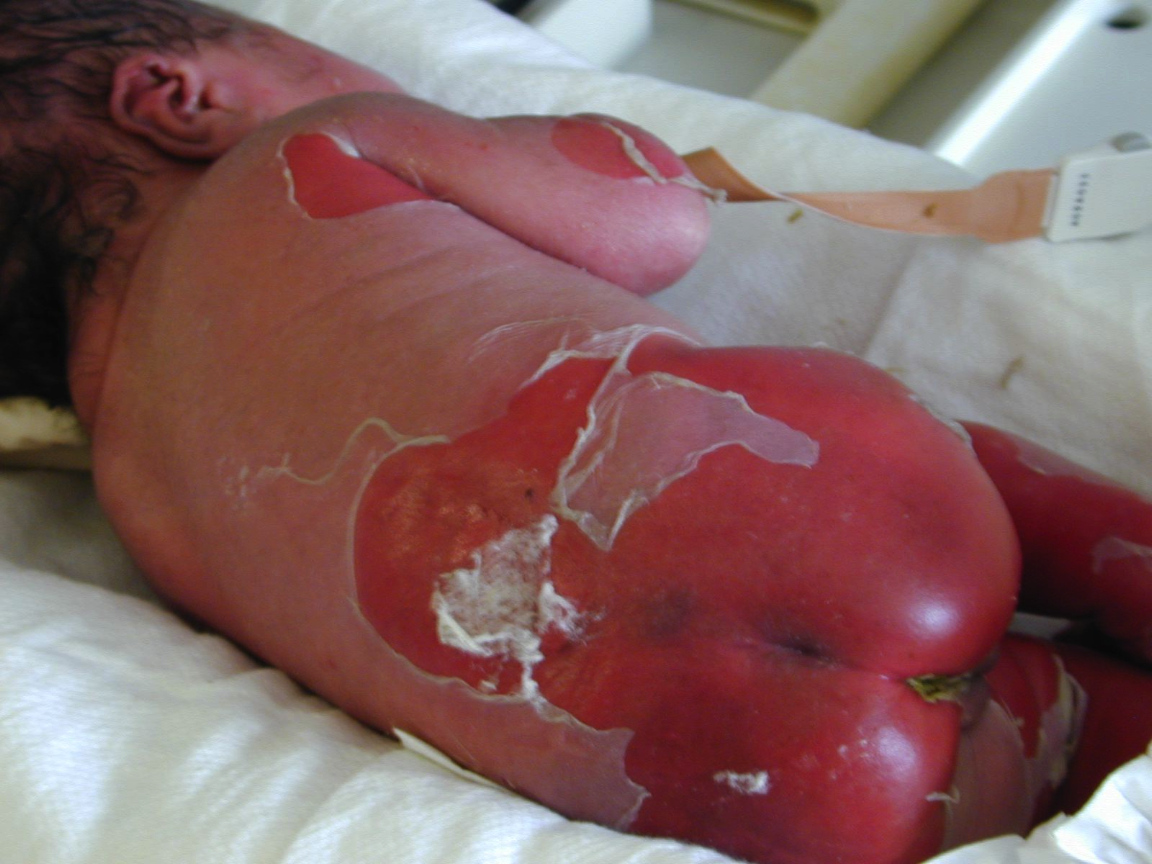
| Figure 6. Our patient with BCIE at birth. Note, the widespread erythema and desquamation. |
Similar to EBS, BCIE also presents with widespread blistering in the newborn period. BCIE is an autosomal dominant disorder, whose initial neonatal presentation with bullae, erythema, and desquamation (Fig. 6) is replaced in early childhood by ichthyosis, scaling, and hyperkeratosis, which persists for the remainder of the patient's life [3, 19]. During infancy, serious potential complications including sepsis and fluid and electrolyte imbalances may arise [3]. After the first few months, however, the erythema and bullae are replaced with generalized hyperkeratosis and dark brown to gray-white scale, especially over joints, resulting in a characteristic cobblestone-like appearance [3, 19]. Verrucous hyperkeratosis is most notable on the palms, soles, and over the flexor surfaces. Hair, nails, and mucosal surfaces are typically uninvolved [3]. Furthermore, the skin generally emits a pungent odor, likely due to bacterial superinfection of macerated skin [3]. It is also important to note the high frequency of spontaneous mutation;approximately one-half of cases, including our patient, have no family history of BCIE [3, 19]. Furthermore, it is also important to screen individuals with verrucous epidermal nevi and biopsy these lesions for BCIE changes because many of these lesions represent a localized form of BCIE secondary to a postzygotic somatic mutation. If this is present in the gonads, it can manifest in the offspring as generalized BCIE [20, 21, 22].
The mutations leading to BCIE result from alterations in genes encoding keratin K1 and K10; however, the phenotypic appearance may differ substantially [3, 19]. Six distinct phenotypes of BCIE have been described [19]. BCIE can first be divided into two groups based on the presence or absence of palmar and plantar involvement. The palmoplantar (PS) type of BCIE is characterized by palm and sole hyperkeratosis, predominantly associated with a keratin 1 mutation. Any potential disturbance of palmoplantar skin from a mutation in keratin 10 is believed to be offset by keratin 9, a protein specific to palmoplantar skin that is thought to complex with keratin 1 and by doing so partially compensate for keratin 10 associated cytoskeletal disruption [23]. The PS group can be further divided into 3 subtypes: PS-1, PS-2 and PS-3. The BCIE type that spares the palms and soles (NSP) can also be further divided into three subtypes: NPS-1, NPS-2 and NPS-3. Subdivisions of the PS and NSP subtypes are based on further characterization of clinical features such as surface characteristics of palmar and plantar skin, presence of contractures, type and degree of scaling, distribution of scaling, and the presence or absence of erythroderma and blistering. Our patient with severely thickened bilateral palmar and plantar hyperkeratosis, "dirty appearing" tan scale, generalized distribution of scaling, neonatal-only incidence of blistering, and absence of erythroderma appears to best fit into the PS-3 subtype of BCIE [24].
 |
| Figure 7 |
|---|
| Figure 7. Punch biopsy of BCIE lesion at 4 years demonstrates hyperkeratosis with vacuolar degeration in the upper epidermis leading to vacuole formation. Irregular keratohyaline granules and acantholysis are also prominent findings. |
In contrast to EBS where clefting occurs in the basal cell layer of the epidermis corresponding to the location of keratins 5 and 14, BCIE is a disorder of keratins 1 and 10 which are key components of the keratinocytes residing higher up in the epidermis, namely the spinous and granular cell layers. These differences in keratin composition of keratinocytes result from the normal maturation and differentiation of keratinocytes with upregulation of K1 and K10 synthesis and downregulation of K5 and K14 synthesis as the keratinocytes migrate away from the basal cell layer of the epidermis [13]. Furthermore, normal expression of keratin 10 is associated with inhibition of keratinocyte proliferation, and hence disruption or loss of normal K10 function not only disrupts the integrity of the keratinocyte cytoskeleton predisposing the epidermis to cytolysis and blister formation, but also predisposes the epidermis to a hyperproliferative state [25]. Early microscopic features of BCIE include keratin intermediate filament aggregation or clumping at the suprabasilar level (seen by electron microscopy) [26] and epidermolysis manifesting as clefting in the spinous or granular cell layers as seen on light or electron microscopy [19]. Later as the infant grows older and his or her condition progresses towards its final state, other histopathologic findings including hyperkeratosis, hypergranulosis with irregularly shaped keratohyaline granules, acantholysis, and areas of suprabasilar epidermolysis often containing characteristic epidermal cells with perinuclear vacuolization with indistinct peripheral boundaries can also be appreciated (Fig. 7) [3, 19]. Similar to EBS, indirect immunofluorescence examination using monoclonal antibodies for keratin expression may allow determination of the specific keratin type involved, allowing for a more precise diagnosis [3]. Furthermore, like EBS, genetic testing and prenatal diagnosis are also commercially available (http://www.genedx.com) [27, 28].
Treatment for BCIE is primarily symptomatic as there is no definitive cure. A regimen of daily bathing with application of an emollient twice daily is essential [3]. Treatment with alpha-hydroxy acids and polyhydroxy-acids, which normalize keratinization, may reduce symptoms of dryness and improve cosmetic appearance with consistent use. Regular shampooing and gentle debridement may reduce scalp scaling. For severe cases, acitretin may be beneficial and doses of 0.5-1.0 mg/kg body weight have reportedly been effective in reducing hyperkeratosis [3]. Though they aid in desquamation, systemic retinoids should be used with caution as they may exacerbate blister formation [3, 6].
 |
| Figure 8 |
|---|
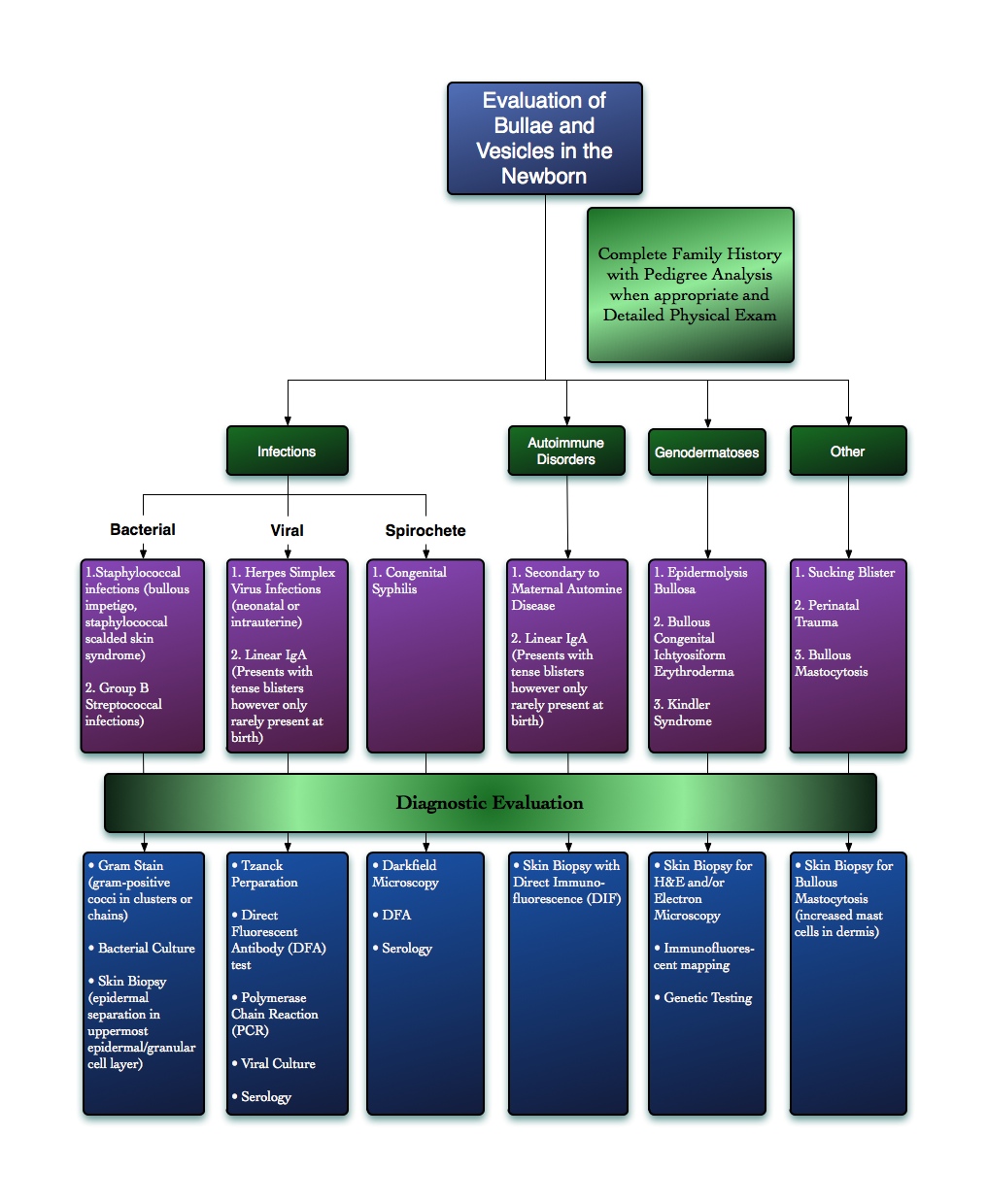
| Figure 8. Algorithm for evaluating blisters and vesicles in the newborn; adapted from Dermatology by Bolognia, Rapini, and Jorizzo [29] |
Table 1 delineates general characteristics of EBS and BCIE, emphasizing similarities and differences. BCIE can be a difficult diagnosis to make in the newborn, especially since 50 percent or more of cases are sporadic and some cases can arise in offspring of individuals with undiagnosed somatic mutation mascarading as epidermal nevi. The differential diagnosis is quite extensive as shown in Figure 8. The workup of blistering disorders in the newborn should include a thorough physical exam and family history. An infectious or autoimmune etiology should be ruled out with appropriate testing and a skin biopsy should be obtained to further evaluate histopathologic features of the epidermis and location of blister formation. The biopsy specimen can be cut into the appropriate number of pieces for routine hematoxylin and eosin staining and/or evaluation by electron microscopy when available. For cases that are difficult to diagnose or when further confirmation of defects in specific proteins and genes is needed, immunoflourescence mapping and genetic testing are also available and should be used to facilitate proper diagnosis.
It is important to make an early diagnosis, preferably in the newborn period, in order to avoid delay of appropriate monitoring and treatment. If the initial diagnosis is missed, the patient will start to develop new clinical features more consistent with an ichthyotic skin disorder in place of the extensive blistering, erosions and erythema so characteristic of the newborn period. With the new hyperkeratotic skin findings also comes a new set of disorders to be considered and ruled out as the differential diagnosis shifts to one that predominantly includes other keratinization disorders and ichthyoses [3]. However, if an observant clinician can pick up on this unique transition between a primarily bullous disorder in the neonate to one of hyperkeratosis in early childhood then the diagnosis can be more easily determined and can be further confirmed with additional testing. Importantly though, and despite the broad differential of vesiculo-bullous disorders in the neonate, with careful evaluation it is possible to make an accurate and early diagnosis of BCIE prior to this transitional state and with an early diagnosis, allow patients to enjoy appropriate and timely treatment without the complications and frustration that accompany an incorrect diagnosis.
References
1. Freeman EB, Koglmeier J, Martinez AE, Mellerio JE, Haynes L, Sebire NJ, et al. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol. 2008 Jun;158(6):1308-14. [PubMed]2. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008 Jun;58(6):931-50. [PubMed]
3. Lacz NL, Schwartz RA, Kihiczak G. Epidermolytic hyperkeratosis: a keratin 1 or 10 mutational event. Int J Dermatol. 2005 Jan;44(1):1-6. [PubMed]
4. Milstone LM, McGuire J, Ablow RC. Premature epiphyseal closure in a child receiving oral 13-cis-retinoic acid. J Am Acad Dermatol. 1982 Nov;7(5):663-6. [PubMed]
5. Baden HP, Buxman MM, Weinstein GD, Yoder FW. Treatment of ichthyosis with isotretinoin. J Am Acad Dermatol. 1982 Apr;6(4 Pt 2 Suppl):716-20. [PubMed]
6. Kwak J, Maverakis E. Epidermolytic hyperkeratosis. Dermatol Online J. 2006;12(5):6. [PubMed]
7. Lawson JP, McGuire J. The spectrum of skeletal changes associated with long-term administration of 13-cis-retinoic acid. Skeletal Radiol. 1987;16(2):91-7. [PubMed]
8. Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005 Jan-Feb;23(1):33-40. [PubMed]
9. Horn HM, Tidman MJ. The clinical spectrum of epidermolysis bullosa simplex. Br J Dermatol. 2000 Mar;142(3):468-72. [PubMed]
10. McGrath JA, Ishida-Yamamoto A, Tidman MJ, Heagerty AH, Schofield OM, Eady RA. Epidermolysis bullosa simplex (Dowling-Meara). A clinicopathological review. Br J Dermatol. 1992 May;126(5):421-30. [PubMed]
11. Dowling GB, Meara RH. Epidermolysis bullosa resembling juvenile dermatitis herpetiformis. Br J Dermatol. 1954 Apr;66(4):139-43. [PubMed]
12. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009 Feb;60(2):203-11. [PubMed]
13. Lane EB, McLean WH. Keratins and skin disorders. J Pathol. 2004 Nov;204(4):355-66. [PubMed]
14. Bergman R, Harel A, Sprecher E. Dyskeratosis as a histologic feature in epidermolysis bullosa simplex-Dowling Meara. J Am Acad Dermatol. 2007 Sep;57(3):463-6. [PubMed]
15. Yiasemides E, Walton J, Marr P, Villanueva EV, Murrell DF. A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. American J Dermatopathol. 2006 Oct;28(5):387-94. [PubMed]
16. Rugg EL, Baty D, Shemanko CS, Magee G, Polak S, Bergman R, et al. DNA based prenatal testing for the skin blistering disorder epidermolysis bullosa simplex. Prenat Diagn. 2000 May;20(5):371-7. [PubMed]
17. Rugg EL, Horn HM, Smith FJ, Wilson NJ, Hill AJ, Magee GJ, et al. Epidermolysis bullosa simplex in Scotland caused by a spectrum of keratin mutations. J Invest Dermatol. 2007 Mar;127(3):574-80. [PubMed]
18. Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009 Jan;34(1):20-5. [PubMed]
19. Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson-Bostom L. Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol. 2008 Jul;59(1):86-90. [PubMed]
20. Paller AS. Laboratory tests for ichthyosis. Dermatol Clin. 1994 Jan;12(1):99-107. [PubMed]
21. Reddy BS, Thadeus J, Kumar SK, Jaishanker T, Garg BR. Generalized epidermolytic hyperkeratosis in a child born to a parent with systematized epidermolytic linear epidermal nevus. Int J Dermatol. 1997 Mar;36(3):198-200. [PubMed]
22. Paller AS. Expanding our concepts of mosaic disorders of skin. Arch Dermatol. 2001 Sep;137(9):1236-8. [PubMed]
23. Smith F. The molecular genetics of keratin disorders. Am J Clin Dermatol. 2003;4(5):347-64. [PubMed]
24. DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol. 1994 Aug;130(8):1026-35. [PubMed]
25. Paramio JM, Casanova ML, Segrelles C, Mittnacht S, Lane EB, Jorcano JL. Modulation of cell proliferation by cytokeratins K10 and K16. Mol Cell Biol. 1999 Apr;19(4):3086-94. [PubMed]
26. Ishida-Yamamoto A, Takahashi H, Iizuka H. Immunoelectron microscopy links molecules and morphology in the studies of keratinization. Eur J Dermatol. 2000 Aug;10(6):429-35. [PubMed]
27. Rothnagel JA, Lin MT, Longley MA, Holder RA, Hazen PG, Levy ML, et al. Prenatal diagnosis for keratin mutations to exclude transmission of epidermolytic hyperkeratosis. Prenat Diagn. 1998 Aug;18(8):826-30. [PubMed]
28. Tsuji-Abe Y, Akiyama M, Nakamura H, Takizawa Y, Sawamura D, Matsunaga K, et al. DNA-based prenatal exclusion of bullous congenital ichthyosiform erythroderma at the early stage, 10 to 11 weeks' of pregnancy, in two consequent siblings. J Am Acad Dermatol. 2004 Dec;51(6):1008-11. [PubMed]
29. Bolognia JL, Rapini RP, Jorizzo JL. Dermatology. London ; New York: Mosby 2003.
© 2009 Dermatology Online Journal