Lipoid proteinosis in two siblings: A report from south India
Published Web Location
https://doi.org/10.5070/D39v02h90cMain Content
Lipoid proteinosis in two siblings: A report from south India
KN Shivaswamy 1, Devinder M Thappa1, Chandrashekar Laxmisha1, and S Jayanthi2
Dermatology Online Journal 9 (5): 12
From the Departments of Dermatology and STD (1), and Pathology (2), JIPMER, Pondicherry - 605 006, India. dmthappa@jipmer.edu
Abstract
A 6-year-old girl and her 9-year-old brother, born of nonconsanguineous parents, had hoarseness and multiple, asymptomatic, raised skin lesions present since childhood. On examination, both siblings had hoarseness and numerous skin-colored, waxy papules distributed over the forehead, face, neck, axilla, groin, and extremities. Acneiform (pocklike) scars were present on the face, trunk, and extremities. Eyelid beading (moniliform blepharosis) was present over bilateral upper and lower eyelids. The tongue, lips, and frenulum were thickened and infiltrated, and the patients were unable to protrude the tongue out of the mouth. The scalp had patchy alopecia. Histological examination of representative skin specimens (from both siblings) showed deposition of pink, amorphous material in the papillary dermis, around blood vessels, and around appendages. These deposits stained positive with Periodic Acid-Schiff stain, were diastase resistant, and were negative for Congo red, confirming our clinical diagnosis of lipoid proteinosis. Over 250 cases of this rare disorder have been described in the literature, but occurrence of lipoid proteinosis in siblings is rare.
Introduction
Lipoid proteinosis (LP), also known as Urbach-Weithe disease and hyalinosis cutis et mucosae (OMIM 247100), is a rare, autosomal-recessively inherited disorder, characterized by hoarseness from early infancy, together with various cutaneous manifestations (acneiform scarring, waxy papules, eyelid beading, etc.) and noncutaneous manifestations attributed to infiltration of hyaline-like material in the skin, larynx, and various organs [1]. This hyaline material is Periodic Acid-Schiff (PAS) positive and diastase resistant and is believed to be the result of deposition of noncollagenous proteins and glycoproteins [2]. The exact pathogenesis of LP is unknown and is found to be associated with disturbance of collagen metabolism with decrease in type-1 collagen and an increase in type-4 collagen, which deposits in dermis and around blood vessels and appendages [3]. Over 250 cases of this rare disorder have been described [3], but occurrence of lipoid proteinosis in siblings is very rare [4, 5, 6, 7] We herewith are reporting this entity in two siblings from south India.
Case report
Two siblings, a 6-year-old girl and a 9-year-old boy born of nonconsanguineous parents, came to us with hoarse voices and multiple, asymptomatic, raised skin lesions present since childhood. On history, both had normal outcome at birth but started developing hoarseness of the voice (as noticed by their mother) by the end of the first year. Subsequently, small blisters developed over the face, trunk and extremities; these lesions ruptured and healed with scarring. Soon after, they developed waxy eruptions over face, neck, and extremities. There was also a history of hair loss. There was no history of photosensitivity, respiratory obstruction, or epilepsy. No other family member had similar disorder.
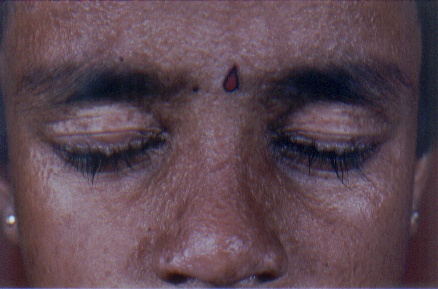
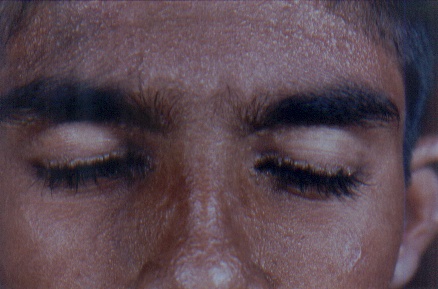
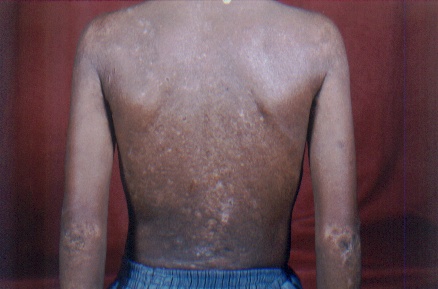
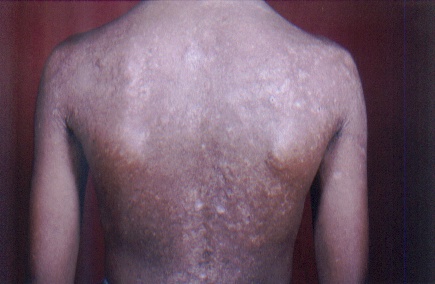
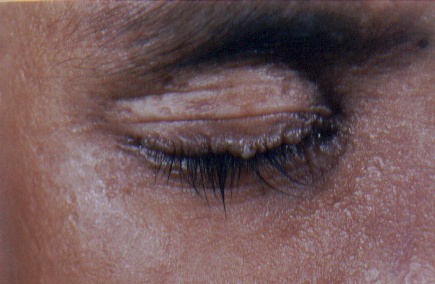
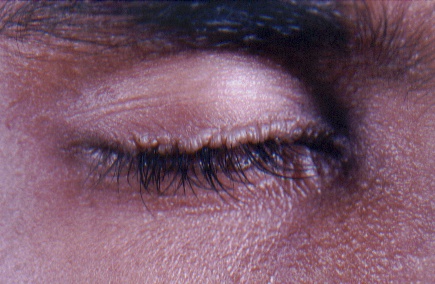
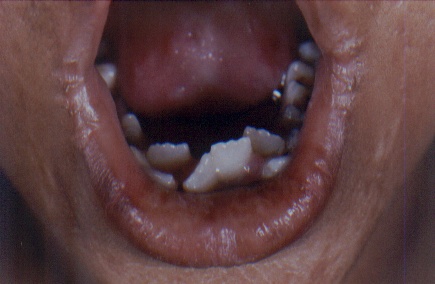
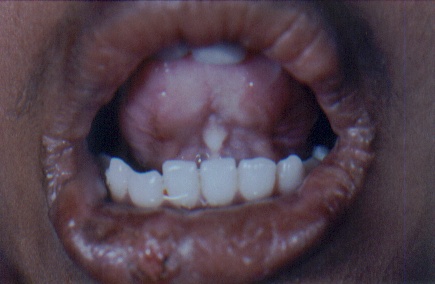
On examination, both the siblings had hoarse voices and numerous, skin-colored, waxy papules on the forehead, face (figs. 1 and 2), neck, axilla, groin, and extremities. Acneiform (pocklike) scarring was evident over face, trunk (figs. 3 and 4), and extremities. Eyelid beading (moniliform blepharosis) was present bilaterally on the upper and lower eyelids (figs. 5 and 6). The tongue, lips, and frenulum were thickened and infiltrated, and there was an inability to protrude the tongue out of the mouth (figs. 7 and 8). Patchy areas of alopecia were present on the scalp. The remainder of the systemic examination including that of central nervous system was found to be normal.
|
|
| Figure 1 | Figure 2 |
|---|---|
| Waxy papular eruption over the face with pock like marks in the female and male siblings. | |
|
|
| Figure 3 | Figure 4 |
|---|---|
| Infiltrated skin of the trunk and upper extremities with numerous scars. | |
|
|
| Figure 5 | Figure 6 |
|---|---|
| Beaded eyelid margins in both siblings. | |
|
|
| Figure 7 | Figure 8 |
|---|---|
| Infiltrated lips with woody indurated tongue and pearly deposits in the frenulum and angles of the mouth. | |
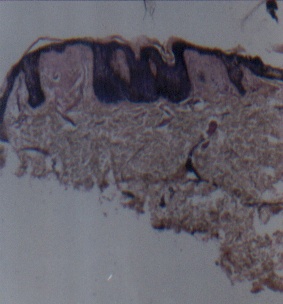
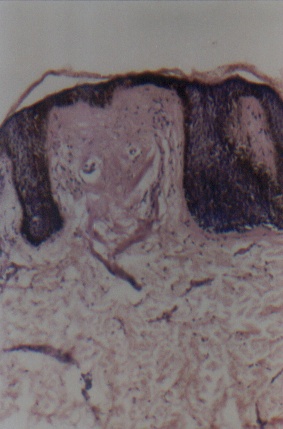
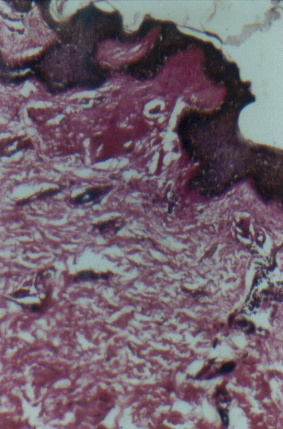
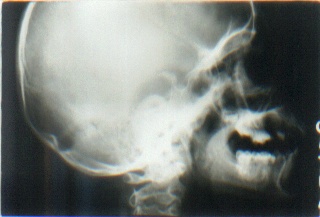
Routine hematological and biochemical investigations were within normal limits. Histological examinations of representative skin lesions from both siblings showed deposition of a pink, amorphous material in the papillary dermis, and around blood vessels and appendages (figs. 9 and 10). These deposits stained positive with PAS stain, were diastase resistant (fig. 11), and negative with Congo red stain. Skull radiography revealed bean-shaped calcification in the suprasellar area of the boy (fig. 12) but was normal in the girl.
|
|
| Figure 9 | Figure 10 |
|---|---|
| Photomicrograph demonstrating hyperkeratosis, acanthosis, and thinning of epidermis overlying the papillary dermal pale eosinophilic deposit (H&E x 40) (fig. 9). Higher magnification photomicrograph showing pale eosinophilic amorphous material in the dermal papilla and thinning of overlying epidermis (H&E x 100) (fig. 10). | |
|
|
| Figure 11 | Figure 12 |
|---|---|
| Photomicrograph showing PAS stained material in the papillary dermis with duplication of basement membrane (PAS x 100) (fig. 11). An X-ray of the skull lateral view shows bean-shaped calcification in the suprasellar area of male sibling (fig. 12). | |
Discussion
Lipoid proteinosis (LP) is a rare, recessively inherited disorder, characterized by deposition of hyaline-like material in the skin, larynx, and other organs, leading to hoarseness from early infancy, and protean manifestations [1]. Although lipoid proteinosis is most prevalent among descendents of German immigrants to South Africa, it is seen worldwide.
The exact pathogenesis of this disease is not known but has been postulated to be the result of either a lysosomal storage disorder involving multiple enzyme defects or from a disturbance in collagen synthesis, as evidenced by decrease in the ratio of type-1 to type-3 collagen associated with a decrease in mRNA for type-1 procollagen. There is also an increase in mRNA for type-4 procollagen resulting in underproduction of fibrous collagens and an overproduction of basement membrane collagens, which tend to deposit in the skin and various organs, the hallmark of the disease [3, 8, 9]. Recent studies have shown that LP is the result of reduced expression of exatracellular-matrix-protein (ECM-1) gene (composed of two alternatively spliced isoforms, ECM-1a and ECM-1b, the latter lacking exon 7 of this 10-exon gene) mapped to chromosome 1 in the fibroblasts [3, 10]. Extracellular-matrix protein 1, a glycoprotein expressed in several tissues including skin, has important physiological and biological roles in epidermal differentiation, in binding of dermal collagens and proteoglycans, and in regulation of angiogenesis. To date, pathogenic mutations have been described in sixteen different families with lipoid proteinosis throughout the world. Homozygous-nonsense mutation in exon 2 of the ECM1 gene, Q32X, was identified by direct sequencing of genomic DNA of an affected member of a Libyan family [11].
The protean manifestations of LP are associated with the extensive deposition of noncollagenous proteins and glycoproteins in the skin, larynx, and other organs [12, 13]. Hoarseness may develop either in infancy or during childhood, and this symptom is usually the earliest feature of LP.
Skin lesions follow hoarseness, including waxy papules on the face, trunk, flexures, and extremities. Acneiform or pocklike scarring may occur predominantly on face and trunk. Usually in the older age group, hyperkeratotic or verrucous lesions resembling xanthoma may be seen on elbows, knees, and buttocks. Such lesions were absent in our siblings, probably because of younger age [12]. Infiltration of the eyelids gives rise to beaded appearance (moniliform blepharosis), which is characteristic for this condition [12, 14]. Infiltration of the oral cavity leads to thickened, wood-hard tongue along with thickened lips and frenulum resulting in inability of the tongue to protrude out of the mouth. Poor dental hygiene may result from dryness of mouth associated with infiltration and obstruction of the parotid duct. Extracutaneous features may include epilepsy, mental retardation, and other neuropsychiatric illnesses. In a few cases, calcifications of the temporal lobe or hippocampi appear as bean-shaped opacities on skull radiography and are considered to be pathognomonic of the disease, as is hoarseness and eyelid beading [15].
Histologically, LP is characterized by deposition of PAS-positive, diastase-resistant material at the level of the basement membrane (resulting in its thickening at the dermoepidermal junction), papillary dermis, surrounding blood vessels, and around adnexal epithelia especially sweat coils [2, 16]. Ultrastructural examination reveals concentric rings of excess basement membrane surrounding blood vessels, and irregular reduplication of lamina densa at dermoepidermal junction resulting in onion-skin appearance [8]. Biochemically, this material is characterized by decrease in type-1 collagen with overproduction of type-4 or basement-membrane collagen. The hyaline deposits in the biopsies examined consist of a carbohydrate-protein complex containing hyaluronic acid and probably chondroitan-sulphate, plus large amounts of lipids [16].
LP needs to be differentiated from erythropoietic protoporphyria (EPP), a condition characterized by skin involvement confined to sun-exposed areas, and associated with photosensitivity. EPP also has a deposition of PAS-positive material, however it is less dense around blood vessels and never occurs around sweat coils. LP can also be differentiated histologically from amyloidosis and xanthomas, two other diseases associated with deposition of material in the eyelids [2, 17, 18].
Treatment of this condition is usually unsatisfactory. Reported approaches include oral steroids, dimethyl sulphoxide [17], intralesional heparin, and etretinate [3]. CO2 laser surgery of vocal cords and beaded eyelid papules, and dermabrasion of skin result in cosmetic improvement. Except for the respiratory obstruction that occurs infrequently and rarely requires tracheostomy, life expectancy is usually normal [3].
References
1. Black MM. Lipoid Proteinosis; Metabolic and nutritional disorders. In: Champion RH, Burton JL, Burns DA. Breathnach SM, eds. Rook/Wilkinson/Ebling Textbook of Dermatology, 6th edn, Oxford: Blackwell Science. 1998: 2460-2462.2. Touart DM, Sau P. Cutaneous deposition diseases. Part1. J Am Acad Dermatol 1998; 39:149-171.
3. Hamada T, Lipoid proteinosis. Clin Exp Dermatol 2002; 27:624-629.
4. Sethuraman G, Tejasvi T, Khaitan BK, Handa KK, Rao S, Singh MK, Sirka C, Sharma VK. Lipoid Proteinosis in Two Siblings: A Report from India. J Dermatol 2003; 30: 562-565.
5. Nanda A, Alsaleh QA, Al-Sabah H, Ali AM, Anim JT. Lipoid proteinosis: report of four siblings and brief review of the literature. Pediatr Dermatol. 2001 ;18: 21-26.
6. Bohme M, Wahlgren CF. Lipoid proteinosis in three children. Acta Paediatr. 1996 ;85:1003-1005.
7. Oezarmagan G, Baykal C, Gursoy EO, Yilmazer S, Buyukbabani N, Coban O. Lipoid proteinosis in 2 sisters. Hautarzt. 1993 ;44: 315-318.
8. Moy LS, Moy RL, Matsuoka LY, Ohta A, Uitto J. Lipoid proteinosis: Ultrastructural and biochemical studies. J Am Acad Dermatol 1987; 16:1193-1201.
9. Newton JA, Rasbridge S, Temple A, Pope FM, Black MM, McKee P. Lipoid proteinosis-new immunopathological observations. Clin Exp Dermatol 1991; 16:350-354.
10. Hamada T, Wessagowit V, South AP, Ashton GH, Chan I, Oyama N, Siriwattana A, Jewhasuchin P, Charuwichitratana S, Thappa DM, Lenane P, Krafchik B, Kulthanan K, Shimizu H, Kaya TI, Erdal ME, Paradisi M, Paller AS, Seishima M, Hashimoto T, McGrath JA. Extracellular Matrix Protein 1 Gene (ECM1) Mutations in Lipoid Proteinosis and Genotype-Phenotype Correlation. J Invest Dermatol 2003; 120:345-350.
11. Chan I, El-Zurghany A, Zendah B, Benghazil M, Oyama N, Hamada T, McGrath JA. Molecular basis of lipoid proteinosis in a Libyan family. Clin Exp Dermatol. 2003 ;28: 545-548.
12. Nagasaka T, Tanaka M, Ito D, Tanaka K, Shimizu H. Protean manifestations of lipoid proteinosis in a 16 year old boy. Clin Exp Dermatol 1999; 25:30-32.
13. Karthikeyan K, Thappa DM, Rakhesh SV. Spot the diagnosis. J Postgrad Med 2002; 48: 210 & 212.
14. Thappa DM, Gupta S. Eyelid beading-A useful diagnostic clue for lipoid proteinosis (Images in clinical practice). Indian Pediatr 2001; 38: 97.
15. Ramsey ML, Tschen JA, Wolf JE. Lipoid proteinosis. Int J Dermatol 1985; 24:230-232.
16. Harper JI, Filipe MI, Staughton RCD. Lipoid protenosis: Variations in histochemical characteristics. Clin Exp Dermatol 1983; 8: 135-141.
17. Wong CK, Lin CS. Remarkable response to oral dimethyl sulphoxide. Br J Dermatol 1998; 119:541-544.
18. Bozdag KE, Gul Y, Karaman A. Lipoid proteinosis. Int J Dermatol 2000; 39:203-204.
© 2003 Dermatology Online Journal