The spectrum of cutaneous reactions associated with calcium antagonists: A review of the literature and the possible etiopathogenic mechanisms
Published Web Location
https://doi.org/10.5070/D39h3065pbMain Content
The spectrum of cutaneous reactions associated with calcium antagonists: A review of the literature and the possible etiopathogenic
mechanisms
Palamaras Ioulios, Michaelides Charalampos, and Tsele Efrossini
Dermatology Online Journal 9 (5): 6
Dermatology Department, West Attika General Hospital of Athens, Greece. ioulios@hol.gr
Abstract
Calcium antagonists (CAs) or calcium-channel blockers, are a common group of antihypertensive medications. These drugs have the property of blocking the calcium channels of the vascular and cardiac smooth-muscle fibers. They have been associated with cutaneous reactions ranging from exanthems to severe adverse events. The frequency of these reactions may be as high as 48 percent. The most common are ankle or pedal edema (up to 30 %), gingival hyperplasia (up to 21 %), and flushing (up to 10 %). Less common are facial or truncal telangiectasia, photosensitivity reactions, new-onset psoriasis (as well as exacerbation of it), purpuric exanthems, pemphigoid manifestations, subacute cutaneous lupus erythematosus, gynecomastia, erythromelalgia, and oral ulcers. Particular adverse manifestations relate to drug potency, degree of vasodilatation, patient age, coexistence of other diseases, co-administration of other cytochrome P450 CYP3A-metabolized medications, fibroblast stimulation, and blood cell effects. Calcium antagonists are associated with a wide range of skin reactions, and the dermatologist should include these in the differential diagnosis of cutaneous diseases.
Introduction
The drugs classified as calcium antagonists (CAs), or calcium-channel blockers (CCBs) are a diverse group of antihypertensive medications introduced into clinical medicine in the 1960s [1]. They possess the property of blocking the transmembrane flow of calcium ions into the cell through voltage-gated channels [1], thus inhibiting the activation of the actin-myosin complex and muscular contraction [2].
At the present time there are six known different calcium-channel subtypes (see table 1) [2]. The L-type has been found in cardiac muscle, vascular smooth muscle (SM) (arterial and venous), nonvascular SM (bronchial, gastrointestinal, genitourinary, uterine), and noncontractile tissues (pancreas, pituitary, adrenal glands, salivary glands, gastric mucosa, white cells, platelets, and lacrimal tissue) [1].
The structure of the voltage-sensitive calcium channel comprises the ion-conduction pore α1-subunit (through which calcium ions pass) and several accessory subunits designated α2, β, γ, and δ. Variations in the α1-subunit account for the differences in voltage and pharmacologic sensitivity between the voltage-sensitive channel subtypes [2]. The α1-subunit itself contains several different functional regions to which the CAs bind, with CAs of different classes (see below) binding to different regions [1]. The α2-, β-, γ-, and δ- subunits modulate the function of the α1-subunit. Regulation of the entire calcium channel itself is a function of catecholamines, angiotensin II, endothelin, and hormones[1].
Pharmacologically, CAs are divided into benzothiazepines (diltiazem), phenylalkylamines (verapamil), dihydropyridines (nifedipine), and tetraols (mibefradil)[2].
The current, most useful classification is the one based on receptor binding properties, tissue selectivity, and pharmacokinetic profiles proposed by Luscher and Cosentino (see table 2) [3].
The CAs, which are currently used for the treatment of hypertension, angina, supraventricular arrhythmias, and in one case for the prevention and short-term therapy of neurological deficits from subarachnoid hemorrhage, are amlodipine besilate, barnidipine, bepridil cilnidipine, diltiazem hydrochloride, efonidipine felodipine, fendilline, isradipine, lacidipine, lercanidipine, mibefiadil, nicardipine nimodipine, nisoldipine, nitrendipine, nifedipine, and nilvadipine. Only diltiazem hydrochloride, verapamil, nifedipine, and nicardipine are available in intravenous formulation.
CAs have also been used in dermatology for the treatment of erythromelalgia (nifedipine extended release-ER, diltiazem) [4], calcinosis in juvenile DM (diltiazem) [5], Raynaud's phenomenon in systemic sclerosis (nifedipine, diltiazem, amlodipine, isradipine, nisoldipine, and felodipine) [6, 7, 8, 9], perniosis (nifedipine) [10], chronic anal fissures (oral or topical use of diltiazem gel) [11], pain relief in postherpetic neuralgia by iontophoresis (nicardipine, verapamil, and diltiazem) [12, 13], keloids (intralesional verapamil) [14], and for the prevention of skin-flap necrosis in rats and rabbits (nifedipine, verapamil [15, 16], nimodipine [17], nitrendipine, and diltiazem [18]) by topical and systemic administration.
Pharmacodynamics
The main effect of CAs is vasodilation with lowering of the blood pressure. Verapamil, gallopamil, diltiazem, bepridil, mibefradil, and monatepil also delay atrioventricular conduction and decrease the myocardial contractility. Several CAs inhibit the growth and proliferation of vascular smooth muscle and fibroblasts. They may also inhibit synthesis of extracellular matrix proteins (collagen, fibronectin, and proteoglycans).
Conversely, nifedipine upregulates keratinocyte growth factor (KGF) secretion and gene transcription by gingival fibroblasts in vitro and KGF mRNA transcripts are elevated in drug-induced gingival hyperplasia in vivo [19]. Likewise, gingival fibroblasts, cultured from patients with gingival overgrowth under treatment with nifedipine or nicardipine, give a better cell-proliferation rate, rate of DNA syntheses, and an increased number of epidermal growth factor (EGF) receptors with the use of nifedipine, diltiazem, and verapamil [20]. CAs also have immunomodulatory and dysregulatory effects; in vitro they depress T-cell function and in vivo produce an immune dysfunction by inhibition of potassium efflux in lymphocytes.
Moreover, verapamil inhibits mast-cell degranulation, platelet aggregation, and neutrophil function by calcium-dependent pathways. In addition, nifedipine decreases platelet aggregation [15].
The main adverse effects are related to their pharmacologic action (vasodilation, negative inotropic, and dromotropic action). The vasodilation produced mainly by the dihydropyridines (especially by the immediate-release preparations) appears to be dose related: headache, dizziness, flushing, and tachycardia may result [2]. Constipation may occur with verapamil in over 25 percent of patients [1]. Gingival hyperplasia and peripheral ankle or pedal edema are two unique side effects of CAs that are discussed below [27].
There are several cutaneous reactions associated with CAs (see table 3a, b). The most frequent are flushing, ankle edema, and gingival hyperplasia. Variations in the bioavailability of these drugs appears to be relevant to the pathogenesis of these effects.
Pharmacokinetics
After oral administration, bioavailability of these drugs varies depending on first-pass metabolism in the intestinal wall and liver. CAs are metabolized to less active metabolites, mainly by cytochrome P-450 CYP3A[1]. Thus, bowel disease, liver disease, or age-associated physiologic atrophy of the intestinal villi (and likewise the attenuation of hepatic metabolism and hepatic flow) could decrease the clearance of the drug and provoke an increased CAs effect [1]. Furthermore, hepatic biotransformation of CAs may be greater in women than men [1].
In diabetic patients, the relative risk of adverse events associated with the use of CAs is greater [21]. There are many different mechanisms implicated, and these mechanisms are discussed below. Moreover, we should not overlook the fact that (a) isoforms of cytochrome CYP450 that may form the reactive metabolite are present in the skin, (b) human cultured keratinocytes are able to bioactivate drugs to their reactive metabolites, (c) the well-known photosensitivity of some drugs suggests that they do reach the cutaneous level after systemic administration, (d) the epidermal keratinocyte is a key cell in the initiation and propagation of cutaneous immune reactions; keratinocyte-derived cytokines provide essential signals for the migration of Langerhans cells, therefore antigen presentation and ultimately T-cell activation, and (e) keratinocytes themselves express human leukocyte antigen (HLA-DR) at inflammatory sites, and may serve as antigen presenting cells (APC) in situations that predispose subjects to cutaneous drug reactions [22].
Methods
Presentation of all the spectrum of cutaneous reactions associated with calcium antagonists and their classification according the active substance and frequency appears in tables 3a and 3b.
Results
Flushing
Flushing is a common side effect, caused by vasodilation. The CAs that may elicit this reaction in order of frequency are nifedipine 10.5-25 percent [23, 24], (the percentage is higher for the immediate-release formulation), nisoldipine 7 percent [23], felodipine 4-7 percent [25], isradipine 6 percent (dose related) [26], lacidipine 5 percent [35], nilvadipine 5.8 percent [23], nitrendipine 4.6 percent [23, 29], cilnidipine 4.5 percent [23], barnidipine 3.4 percent [23], nicardipine 1.8-3 percent [23, 26], manidipine, 2.2-2.7 percent [23, 30, 31], amlodipine 1.2-2 percent [32], efonidipine 1.8 percent [23], lercanidipine 1.1 percent [23], diltiazem 0.1-3 percent [23, 33], verapamil < 1 percent [34].
Ankle or pedal edema
Nonvascular peripheral (ankle or pedal) edema has been reported during treatment with almost all CAs; nifedipine 10-30 percent [27] (nifedipine SR: 1-8 %)[24], verapamil 6 percent [27], diltiazem 6-10 percent [27] (diltiazem SR 2-3 %)[33], felodipine 2-17 percent [25], isradipine 6 percent [26,28], amlodipine 6-15 percent (6 % in males, 15 % in females)[32], lacidipine 4-4.4 percent [35, 36], lercanidipine 1.2-9 percent [36, 37], nicardipine 3 percent [26], nisoldipine 6-19 percent [38], manidipine 4.9-6 percent [30, 31], mibefradil 7 percent [39]. This side effect appears to be dose related for the majority of the above enumerated drugs. Concerning the etiopathogenesis of this phenomenon, several mechanisms have been proposed; stimulation of the renin-angiotensin-aldosterone system, fluid-volume retention [40], and precapillary anteriolar vasodilation [41, 42, 43]. CAs increase the vascular-fluid filtration from the intravascular to the extravascular compartment, by mechanisms probably independent of interference with the modulation (relaxation) of the intrinsic myogenic tone of the precapillary arterioles at the dorsum of the foot. This modulation seems to be dose related with these drugs because the newer calcium-channel-blocking agents develop a lower incidence of edema through a balanced pre- and post-capillary vasodilation [41, 43]. Further studies are needed to elucidate the exact pathogenesis of this reaction.
Gingival hyperplasia
Gingival hyperplasia (GH) can be idiopathic, inherited, or iatrogenic. It is characterized by enlarged and occasionally inflamed gums (the interdental papilla and the free gingival margins increase in size and thickness progressively covering the clinical tooth crowns) [44, 45]. The prevalence of overgrowth with the use of CAs may be as high as 38 percent. The incidence is 3.3-times more common in men than in women [46].
To date, the CAs reported to cause GH are verapamil [44], diltiazem [44], nifedipine [47], felodipine [44], isradipine [44], nitrendipine [45], nimodipine [44], amlodipine [48, 49], nicardipine [44, 50], manidipine [51]. The prevalence of nifedipine may be greater than that of other CAs [46]. The incidence may approach 10 percent [24]. The prevalence of amlodipine-related GH was calculated in 3.3 percent [48]. Amlodipine GH associated with facial telangiectasia have also been described in a 3-year-old girl [49]. With felodipine the onset time was 4 weeks after initiation of the treatment. By experimental studies the GH resulted to be dose and time related [44].
Nicardipine [50], (incidence less than 1 %), induced gingival hyperplasia in a 8.5-year-old boy 2 years after the increase of his daily dose from 40 mg to 50 mg.
Histologically, the gingival tissue exhibits hyperplasia of connective tissue with lymphoplasmacytic inflammatory infiltrate. The surface epithelium is acanthotic with the formation of elongated test-tubelike rete ridges [44, 45]. Ultrastructural studies reveal an overproduction of collagen and fibronectin without a significant increase of the area occupied by fibroblasts [52]. Moreover, nifedipine has been found to affect the inflammatory infiltrate with a greater number of B-lymphocytes [53]. There are several studies that propose the etiopathogenesis of this phenomenon. Brown et al. [45] considered a possible defect in collagen degradation. The CAs decrease the uptake of folic acid, which participates in the production of active collagenase enzyme (that degrades collagen) by influencing Ca++/Na+ flux, therefore they inhibit the catabolism of ground substance in the oral mucosa. Nyska et al. [54] suggested a blockage of the aldosterone synthesis in zona glomerulosa of the adrenal cortex by CAs, with subsequent feedback stimulation of an increase in pituitary secretion of ACTH, which produces hyperplasia of zona glomerulosa and increases the secretion of androgen hormones that may act on the gingival cells and matrix to produce gingival hyperplasia. Young et al. [34] and Van der Vlenten [49], proposed that inflammation and gingivitis secondary to bacterial plaque induce the production of gingival crevicular fluid. This serum-derived transudate may cause accumulation of the CA in the gingivae with subsequent localized toxic effect and gingival hyperplasia.
According to the latest data, several cofactors seem to interact with the CAs and influence the onset and severity of this disorder. First, the absorption of the drug and its blood drug level depends on the age of the patient [1, 2, 55]. Second, the gender of the subject if important; males are three times as likely as females to develop gingival overgrowth [56]. Third, the presence of bacterial plaque and gingival inflammation is a major cofactor [56]. Actually, control of gingival inflammation may produce a significant remission [51]. Under conditions of GH, the sequestration of the drug in the crevicular fluid, is up to 290-fold greater than in plasma [57].
Keratinocyte growth factor (KGF) is upregulated by nifedipine. In fact, KGF mRNA transcripts are elevated in drug-induced GH in vivo, and nifedipine upregulates KGF secretion and gene transcription by gingival fibroblasts in vitro[19]. Moreover, CAs stimulate collagenase synthesis in fibroblasts by altering the fibroblast cell shape, enhancing cell-proliferation rate and DNA syntheses, and increasing the number of EGF-receptors (EGF-RCs) [20, 58]. And androgen metabolism and the overproduction of 5-α-dihydrotestosterone (5-α-DHT) and 4-androstenedione upregulate gingival fibroblast proliferation and stimulate matrix synthesis [59]. In fact, a positive correlation between GH and plasma testosterone was noted in dogs whose castration prevents calcium-channel-blocker induced GH [60]. All these factors interact for the development of gingival hyperplasia (see fig. 1).
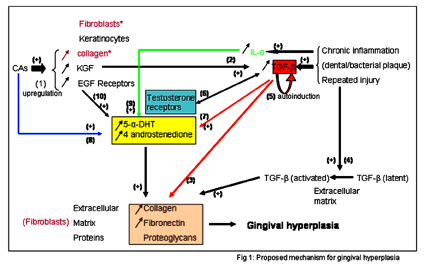
|
| Figure 1 |
|---|
The upregulation of KGF stimulates the keratinocytes. Keratinocytes secrete TGF-β1 [61] (transforming growth factor-β). TGF-β1 has a central role in GH; it stimulates fibroblasts to increase their synthesis of extracellular matrix proteins (i.e. collagen, fibronectin, and proteoglycans). TGF-β1 is also bound locally to the extracellular matrix in latent phase and can be activated after injury. TGF-β1 induces both infiltrating cells and resident cells to produce more of it. This autoinduction amplifies the biological effects of TGF-β1 and with repeated injury, (i.e., the bacterial plaque and chronic gingivitis), the increase in TGF-β1 production is sustained. This creates a chronic vicious circle of TGF-β1 overproduction, which leads to a progressive deposition of extracellular matrix proteins and therefore GH [61]. In addition steroid receptors (such as testosterone receptors), located in the gingivae [60] may act as post-transcriptional regulators for genes of different isoforms of TGF-β1 [61].
Furthermore, TGF-β increases the formation of the biologically effective androgen metabolite 5-α-DHT from testosterone, thus increases the connective-tissue turnover [62]. Moreover, it was found that nifedipine elevates 5-α-DHT and 4-androstenedione metabolism by 24-fold and 32-fold, respectively, in hyperplastic gingival tissue [63]. IL-α stimulates DHT and androstenedione synthesis by 2-fold and EGF, stimulates DHT by 2-fold and testosterone by 5-fold [64]. The importance of these interactions may be seen in the healthy female gingival tissue that normally does not metabolize testosterone significantly. However, in the presence of gingival inflammation, 5-α-DHT formation is comparable to that of male samples [63]. Considering this mechanism, it seems plausible that GH can be likened to the clinical manifestation of a keloid formation in gingival tissue.
Facial telangiectasia
Photodistributed facial telangiectasia has been described for nifedipine [47, 65], felodipine [66], amlodipine [49, 67, 68], and diltiazem [66]. Clinically it is characterized by marked arborizing telangiectasia spreading on all the photoexposed areas of the body: face (more frequently), upper back, shoulders, and extensor aspects of the upper arms, and chest) [68].
Nifedipine-induced telangiectasia was first reported by Tsele et al. [65]. The slow-release formulation is responsible for this effect. There is no gender preference and the onset time is 4-8 months after the initiation of therapy. Usually, within 3-9 months after discontinuation of the drug the telangiectasia is resolving, with gradual dramatic improvement [69]. Felodipine-induced facial telangiectasia is also reported [66]. A considerable improvement was seen 2 months after stopping the drug. With amlodipine [67, 68], the onset of the eruption occurs at up to 12 months, and the remission begins 3 months after withdrawal of the drug, with complete resolution after 6 months.
The etiology of this disorder is not fully understood. Several mechanisms have been postulated: (1) Both the vasodilatory action of the CA and the actinic damage produced in the vessels in photoexposed areas may contribute to this phenomenon [66]. (2) Underlying longstanding untreated rosacea may also be an important factor [66]. (3) Alternatively, it may be an idiosyncratic reaction [69]. (4) An abnormal sensitivity to ultraviolet (UVB and UVA) wave-lengths could generate toxic photoproducts [70]. However, only in vitro and at higher plasma levels (unattainable in vivo), UVA irradiation induces formation of toxic photoproducts by nifedipine [69]. Furthermore, phototesting in vivo has failed to produce consistent results [69]. In addition, photostability and phototoxicity studies with diltiazem have shown that the main photoproduct was diltiazem-S-oxide and the latter was neither mutagenic nor toxic [71]. On the other hand, drug-induced photosensitivity by diltiazem demonstrated a better minimal-erythema-dose response in the UVB region [72].
Telangiectasia related to CAs has also been described on nonphotoexposed areas [73]. Spiderlike telangiectasias were present on the trunk of a 70-year-old woman who had been taking felodipine for several years. Except hypertension, the patient had no other accompanying diseases. It should be emphasized that complete resolution of these lesions was not achieved on stopping the drug. The underlying mechanism remains to be elucidated, but it has been postulated that it might have been resulted by felodipine's chronic vasodilation.
Photosensitivity reactions
These reactions are manifested as generalized or localized erythema or maculopapular rash on the face and upper trunk. They may be induced by nifedipine and diltiazem [47], and there may be a cross-reaction between these agents [74]. The onset time was 5 months after initiating nifedipine and 1 week after diltiazem (on the same patient). Complete clearing occurred within 3 weeks after discontinuation of nifedipine and 1 week after diltiazem. The exact mechanism is still unknown but the perivascular infiltration with mononuclear cells suggests participation of the reticular immune system. It is possible that an antigen formed by a photoreactive drug or metabolite (such as diltiazem-s-oxide [58]) binds to skin proteins and leads to a UV-dependent delayed-hypersensitivity reaction.
A photodistributed hyperpigmentation has been observed in African-American females under treatment with Diltiazem sustained release [75, 76]. Physical examination showed blue-gray reticulated hyperpigmented macules and patches on the photoexposed areas, usually of the upper trunk. The areas involved in order of frequency are face, neck, chest, forearms, and hands. Histologically the changes described consistent of a lichenoid dermatitis with basal vacuolar alteration and prominent pigment incontinence (i.e., lichenoid drug eruption). There are five reported cases of this disorder; In four, the mean duration of diltiazem administration prior to the onset of hyperpigmentation was 8 months. In the fifth case it was 5 years. Over a 50 percent improvement was noticed at 10 months after stopping the drug. UVA is proposed to be a causal agent, and a metabolite may represent the photosensitizer [75].
Other lichenoid eruptions such as lichenoid purpura and widespread lichen planus are described with diltiazem hydrochloride [77] and amlodipine [78], respectively. The first case [77] refers to a 65-year-old male who presented pruritic, purpuric lichenoid papules on his legs and buttocks, 1 month after initiating treatment with diltiazem. Histologically, a lichenoid infiltration of lymphocytes and histiocytes was found into the dermoepidermal junction. A complete remission of the dermatitis was observed 3 weeks after the change of diltiazem. The pathogenic process of lichenoid purpura is believed to be related to immunological events. Diltiazem itself may have played an immunological role (see also pharmacodynamics). The second case [78] is of a 56-year-old female who presented a widespread intensely pruritic lichenoid eruption consisting of violaceous plaques and pigmented macules over the limbs, and the anterior and posterior trunk, two weeks after starting treatment with amlodipine. Wickham striae were also detectable in the oral cavity. There was a significant improvement of the clinical findings, 5 weeks after discontinuation of amlodipine and simultaneous administration of oral acitretin 30 mg daily, oral antihistamines and potent topical corticosteroids.
Psoriasiform eruptions
These eruptions are associated with diltiazem and nifedipine [47]; new-onset psoriasis as well as exacerbations of it, are associated not only with diltiazem [79] and nifedipine [80] but also with felodipine [80] and amlodipine [80]. The median latent period between the precipitation or exacerbation of psoriasis and the beginning of intake of CAs is 28 months. The mean age is 64.1±8.9 years and the male-female ratio is 2.25. Almost half, of the patients are also taking β blockers (the latter as well may cause psoriasis).
There are several presumed mechanisms:
- CAs enhance keratinocyte proliferation and differentiation, which depend on intracellular calcium levels [81, 82].
- β blockers likewise provoke a derangement in calcium metabolism by blocking the calcium influx into the cells; hence, the simultaneous intake of CAs might have a synergistic effect on the onset of psoriasis [80].
- Nifedipine was discovered to inhibit a final event in the cornification of epidermal cells (specifically, the processing of profilaggrin to filaggrin) by blocking the calcium influx into them [80].
Acute generalized exanthematous pustulosis AGEP)
This reaction is associated with acute, extensive formation of sterile pustules, fever (> 38° C) and peripheral blood leukocytosis has been reported with extended-release diltiazem [83, 84] and nifedipine [85]. The clinical and pathological findings are similar to those of pustular psoriasis. The onset time varied from 10-14 days and the eruption improved within 1 week after discontinuation of the drug. Regarding extended-release diltiazem, the patch testing was positive at 2 days.
The etiology remains obscure, however, it is possible that immune-mediated mechanisms may promote the release of potent neutrophil chemoattractants and activators, such as interleukins (ILs) and tumor necrosis factor-α (TNF-α). These mechanisms therefore could be triggered by the CAs or their metabolites, particularly in genetically susceptible individuals [83].
Hypersensitivity syndrome
This type of reaction may be a special risk for patients with psoriasis and concomitant intake of diltiazem [86]. The hypersensitivity syndrome reaction induced by this drug may present not only as an exfoliative dermatitis (rate, cases per million prescriptions = 0.63), but also as a severe toxic epidermal necrolysis (TEN) (rate, cases per million prescriptions = 0.09). CA-induced TEN is characterized by fever and internal organ involvement (hepatic injury, pulmonary, hematologic or renal impairment, and thyroid dysfunction) [79]. Often there is early atypical lymphocytosis and later eosinophilia. The onset of the eruptions varies from 2 to 10 days after initiation of the drug [47]. The etiology of this phenomenon is unknown, but it is supposed to represent an idiosyncratic reaction [86].
Exfoliative dermatitis and Stevens-Johnson-Syndrome/TEN
Exfoliative dermatitis was described with verapamil [79] (rates, cases per million prescriptions = 0.1) and nifedipine [79] (rate = 0.42). Thus, we can observe comparable rates for nifedipine and diltiazem but only a single report cited for verapamil.
Severe adverse reactions such as erythema multiforme (EM), and Stevens-Johnson syndrome (SJS), have been reported with verapamil [79] (rate, cases per million prescriptions=0.21 and 0.65 respectively), diltiazem [79] (0.44 and 0.27), nifedipine [79] (0.07 and 0.15), and amlodipine [87], either with primary treatment or following substitution for nifedipine. As a result, the rate of SJS, TEN (combined), and EM was nearly identical for diltiazem and verapamil [79]. The average age of patients was 64 years. In EM and SJS, the time from initiating medication to reaction onset averaged from 2 days to 3 months [79,88]. With verapamil, the resolution began approximately 4 days after discontinuation of the drug [89].
The underlying mechanisms remain unclear, but it has been proposed that these drugs may be able to form sensitizing haptens [87].
Subacute cutaneous lupus erythematosus (SCLE).
This form of lupus has been associated with verapamil, diltiazem, and nifedipine [90]. The mean patient age was 67 (from 48 to 86 years) and the female-male ratio: 3.5. The time from starting the medication to onset reaction varied from 6 months to 5 years. As to the nine patients reported, 78 percent were antinuclear-antibody (ANA) positive, and 89 percent were anti-Ro- (SSA) or anti-La- (SSB) antibody positive. Two patients had evidence of concomitant or preexisting connective-tissue disease. Complete resolution was observed within 2-12 weeks of drug cessation. ANAs were reduced, but anti-Ro and anti-La remained positive.
As for the etiology, it has been postulated a two-fold mechanism. First, CAs may induce a Ro and La antigen displacement to the surface of the keratinocyte by altering the cytosolic calcium concentration [90, 91, 92]. Second, they provoke a perturbation of lymphocyte function that could create an immunologic environment and therefore allow the emergence of autoantibodies [93, 94, 95]. These may bind to Ro and La antigens and result in keratinocyte injury by complement-mediated lysis and antibody-dependent cellular cytotoxicity (ADCC), similarly in the manner proposed for idiopathic SCLE [96, 97].
Pemphigus and pemphigoid
Pemphigus foliaceous and pemphigoid nodularis have been described with nifedipine [98]. The latter was presented on a 70-year-old male after 3 months of nifedipine 10 mg daily. On physical examination there was evidence of pruritic papules and nodules initially on the upper back and afterward on the trunk, limbs, face, and scalp. Ulceration, which healed without scarring, was present only in the mouth. In addition, involvement of the nail beds of several fingers and toes resulted in the formation of pterygium unguis.
The finding of the skin biopsy and direct immunofluorescence (IMF), supported the diagnosis of nodular pemphigoid. Furthermore, circulating antibodies anti-basement membrane zone (BMZ) staining with IgG and C3 were detected to a titer 1:800 by indirect IMF. Moreover, antibodies to the 230-kDa pemphigoid antigen were found by Western blot. With the cessation of nifedipine, the patient's skin showed considerable improvement, together with a fall in indirect IMF titer to 1:100.
There are several theories that try to explain the etiopathogenesis of drug-induced pemphigoid. Some authors believe that drugs may bind in the lamina lucida and act as haptens inducing the formation of anti-basement-membrane-zone antibodies [99]. Others suggested that drugs may structurally modify molecules and uncover hidden epitopes, thereby stimulating an autoimmune response [100]. It is presumed that skin susceptibility to nifedipine might be genetically determined, with some nifedipine-treated patients developing an acantholytic reaction and others a subepidermal bullous eruption [101].
Erythromelalgia
This reaction has been related to nifedipine [79], diltiazem [47], verapamil [102], and nicardipine [103]. On the other hand, nifedipine extended release and diltiazem have also been used for its treatment [104].
There are three subtypes of erythromelalgia: (a) thrombocytosis and hyperviscosity, (b) microvascular ischemia (vasoconstrictive-reactive hyperemia), which is the most frequent and (c) vasodilatory [105]. Regarding the calcium-channel-blocker-induced erythromelalgia the time lapse between the first dose of the drug and its occurrence varied from 8 weeks to 1 year: the time from discontinuation of the drug to resolution ranged from 1 to 14 days [102]. All blood exams including ANA, rheumatoid factor and a general antoantibody screening resulted negative [102]. This drug-induced erythromelalgia may represent a manifestation of the third subtype (vasodilatory). However, the exact mechanism remains unknown.
Nifedipine and diltiazem may have a therapeutic effect on the second subtype of erythromelalgia, by attenuating the vasoconstriction phase and improving the nutritional capillary flow (by relaxing the precapillary's sphincters)[104, 106, 107].
Gynecomastia
Breast enlargement has been associated with diltiazem [108], verapamil [109], nifedipine [109], and amlodipine [110].
Regarding the etiopathogenesis, two different mechanisms have been proposed: First, verapamil, (the oral formulations), but not dihydropyridine and benzothiazepine CAs may raise serum PRL concentrations [111, 112, 113, 114]. In fact the prevalence of hyperprolactinemia was found to be 8.5 percent. There is an enhancement of PRL secretion possibly resulting from decreasing the hypothalamic generation of dopamine [113]. Moreover testosterone levels were lower in patients on verapamil [111]. Thus, a possible mechanism could be the suppression of gonadotropin-releasing hormone/ thyrotropin-releasing hormone (GnRH/TRH), which stimulates anterior-pituitary hormone secretion of luteinizing hormone (LH), hence the decrease of testosterone's synthesis [115].
Another mechanism might be the decreased cytochrome P450 CYP3A activity produced by CAs (verapamil and diltiazem) [1]. Apparently, there is an increase in serum estradiol concentration by inhibiting cytochrome P450 CYP3A-dependent neutralization of estradiol [116, 117]. This increase results in an increment of PRL secretion proportionate to the degree of estrogenization [118].
Oral ulcers
Ulcers of the oral mucosa have been attributed to verapamil and diltiazem [119]. Clinically they appear as non-indurated extremely painful ulcers with a central erythematous zone and a white halo, situated on the lateral borders of the tongue. They are recalcitrant to any known therapeutic intervention. For diltiazem and verapamil the onset time is 5 months and 1 year, respectively, after the initiation of the treatment. A complete healing of the ulcers occurred from 2 weeks to 1 month after discontinuation of the drugs.
The results of the histopathologic examinations are nonspecific: spongiosis, foci of necrosis, and a mixed (lymphocytes and plasma cell) inflammatory-cell infiltrate. The pathogenesis of drug-induced ulceration is related to immunological or nonimmunological mechanisms. In the immunological route, the drug or any of its components may trigger an immune response, which generates an exaggerated reaction directed at the surface epithelium resulting in ulcers. In the nonimmunological route, the drug may directly stimulate mast cells and lymphocytes to release cytotoxic chemical mediators.
Conclusions
This article represents our views regarding the dermatological adverse reactions related to CAs, after a comprehensive review of the published literature. We have attempted to present an unbiased summary of the available data and point out that the dermatologist should be aware of these reactions during the process of differential diagnosis for skin diseases.
References
1. Abernethy D, Schwartz J, Calcium-Antagonists Drugs, N Engl J Med 341(19):1447-1457, 1999.2. Flynn J, Pasko D, calcium-channel blockers: pharmacology and place in therapy of pediatric hypertension, Pediatr Nephrol. 15:302-316, 2000.
3. Luscher T, Cosentino F, The classification of calcium antagonists and their selection in the treatment of hypertension A Reappraisal, Drugs. Vol 55(4):509-517, 1998.
4. Cohen J, Erythromelalgia: New theories and new therapies, J Am Acad Dermatol. (43):841-847, 2000.
5. Palmieri GM et al, Treatment of calcinosis with diltiazem, Arthritis Rheum 38(11):1646-54, 1995.
6. Sturgill MG, Seibold JR, Rational use of calcium-channel antagonists in Raynaud's phenomenon, Curr Opin Rheumatol 10(6):584-8, 1998.
7. No authors listed, Comparison of sustained-release nifedipine and temperature biofeedback for treatment of primary Raynaud phenomenon.Results from a randimozed clinical trial with 1-year follow up, Arch Intern Med 160(8):1101-8, 2000.
8. General approach to the treatment of scleroderma-I, UpToDate(r), 2002.
9. Steen VD, Treatment of systemic sclerosis, Am J Clin Dermatol 2(5):315-25, 2001.
10. Dowd PM et al, The treatment of chilblains with nifedipine: the results of a pilot study, double-blind placebo-controlled randomized study and a long term open trial, Br J Dermatol. 120(2):267-75, 1989.
11. Jonas M et al, A randomized trial of oral vs. topical diltiazem for chronic anal fissures, Dis Colon Rectum Voll 44(8):1074-78, 2001.
12. Ikebe H et al, The effect of iontophoresis with several Ca channel blockers for PHN patients, Masui 44(3):428-33, 1995.
13. Taniguchi K et al, The effect of calcium channel antagonist administered by iontophoresis on pain threshold, Acta Anaesthesiol Belg 46(2):69-73, 1995.
14. D'Andrea F, Brongo S, Ferraro G, Baroni A, Prevention and treatment of keloids with intralesional verapamil, Dermatology 204(1):60-2, 2002.
15. Weinzweig N, Lukash F, Weinzweig J, Topical and systemic calcium-channel blockers in the prevention and treatment of microvascular spasm in rat epigastric island skin flap model, Ann Plast Surg; 42:320-26, 1999.
16. Komorowsjka timek E, Chen S, Zhang F, Dogan T, Lineaweaver W, Buncke H, Prolonged perivascular use of verapamil or lidocaine decreases skin flap necrosis, Ann Plast Surg;43:283-88, 1999.
17. Björn Stark G, Dorer A, Jaeger K, Narayanan K, The influence of the calcium blocker nimodipine on flap survival, Ann Plast Surg;23:306-9, 1989.
18. Kawabata H, Kenneth K, Serena C, Angus J, McC. O'Brien B, Experience with calcium antagonists nitrendipine, diltiazem and verapamil and _2-agonist salbutamol in salvaging ischemic skin flaps in rabbits, Microsurgery 12:160-3, 1991.
19. Das SJ, Olsen I, keratinocyte growth factor is upregulated by hyperplasia-inducing drug nifedipine, Cytokine; 12(10):1566-9, 2000.
20. Matsumoto H, Noji I, AkimotoY, Fujii A, Comparative study of calcium-channel blockers on cell proliferation, DNA and collagen syntheses, and EGF receptors of cultured gingival fibroblasts derived from human nifedipine, nicardipine and nisoldipine responders, J Oral sci;43(4):261-8, 2001.
21. Pahor M, Kritchevsky S, Zuccala G, Guralnik J, Diabetes and Risk Of adverse events with calcium antagonists, Diabetes Care Vol 21;(1):193-4, 1998.
22. Svensson C, Cowen E, Gaspari A, Cutaneous Drug reactions, Pharmacol Rev 53:357-79, 2000.
23. Saruta T, Current status of calcium antagonists in Japan, Am J Cardiol;82:32R-34R, 1998.
24. Nifedipine: drug information handbook, UpToDate(r), 2002.
25. Felodipine: drug information handbook, UpToDate(r), 2002.
26. Kubota K, Pearse GL, Inman WH, Vasodilation-related adverse events in diltiazem and dihydropyridines calcium antagonists studied by prescription-event monitoring, Eur J Clin Pharmacol; 48(1):1-7, 1995 .
27. calcium-channel blockers Chapter 3, Opie L, Drugs for the heart (4th Ed.), 1995.
28. Chrysant SG, Cohen M, Sustained blood pressure control with controlled-release isradipine, Am J Hypertens; 8(1):87-9, 1995.
29. Wang X, Gong L, Guo J, Wang X, Liu Y, Ye X, Zhang G, Yang P, Parallel comparative trial of amlodipine and nitrendipine monotherapy in patients with essential hypertension, J Hypertens Suppl; 16(4):S43-7, 1998.
30. Cheer S, McClellan K, Manidipine: A review of its use in hypertension, Drugs; 61(12):1777-1799, 2001.
31. Fogari R, Zoppi A, Lusardi P, Mugellini A, Efficacy and tolerability of manidipine hydrochloride in the long term treatment of mild-moderate hypertension. Manidipine efficacy in long-term treatment group, Blood Press Suppl 5:24-8, 1996.
32. Osterloh I, The safety of amlodipine, Am Heart J 118(5 Pt 2):1114-9;discussion 1119-20, 1989.
33. Diltiazem: drug information handbook, UpToDate(r), 2002.
34. Verapamil: drug information handbook, UpToDate(r), 2002.
35. Lindholm L, Tcherdakoff P, Zanchetti A, Safety profile of lacidipine, Drugs;57 Suppl.1:27-9, 1999.
36. Leonetti G, Magnani B, Pessina AC, Rappelli A, Trimarco B, Zanchetti A, Tolerability of long-term treatment with lercanidipine versus amlodipine and lacidipine in elderly hypertensives, Am J Hypertens;15(11):932-40, 2002.
37. Barrios V, Navarro A, Esteras A, Luque M, Romero J, Tamargo J, Prieto L, Carrasco JL, Herranz I, Navarro-Cid J, Ruilope LM, Antihypertensive efficacy and tolerability of lercanidipine in daily clinical practice.The ELYPSE study, Blood Press;11(2):95-100, 2002.
38. Opie LH, Muller FO, Myburg DP, Rosendorff C, Sareli P, Seedat YK, Weich DJ, Luus HG, Efficacy and tolerability of nisoldipine coat-core formulation in the treatment of essential hypertension:The south african multicenter ANCHOR study, Am J Hypertens;10(3):250-60, 1997.
39. Karch FE, Pordy R, Benz JR, Carr A, LundeNM, Marbury T, Tarro JN, Comparative efficacy and tolerability of two long acting calcium antagonists, mibefradil and amlodipine, in essential hypertension. Mibefradil hypertension study group, Clin Ther;19(6):1368-78, 1997.
40. Messerli FH, Vasodilatatory edema: a common side effect of antihypertensive therapy, Curr Cardiol Rep;4(6):479-82, 2002.
41. Pedrinelli R, Dell'Omo G, Melillo E, Mariani M, Amlodipine, enalapril, and dependent leg edema in essential hypertension, Hypertension;35:621-5, 2000.
42. Iabichella ML, Dell'Omo G, Mellilo E, Pedrinelli R, calcium-channel blockers blunt postural cutaneous vasoconstriction in hypertensive patients, Hypertension;29:751-756, 1997.
43. Messerli FH, Calcium antagonists in hypertension: from hemodynamics to outcomes, Am J Hypertens 15(7 Pt 2):94S-97S, 2002.
44. Young P, Turiansky G, Sau P, Liebman M, Benson P, Felodipine induced gingival Hyperplasia, Cutis;62(1):41-43, 1998.
45. Brown RS, Sein P, Corio R, Bottomley WK, Nitrendipine-induced gingival hyperplasia, Oral Surg Oral Med Oral Pathol;70:593-6, 1990.
46. Prisant LM, Herman W, Calcium channel blocker induced gingival overgrowth, J Clin Hypertens (Greenwich);4(4):310-1, 2002.
47. Knowles S, Gupta A, Shear N, The spectrum of cutaneous reactions associated with diltiazem: Three cases and a review of the literature, J Am Acad Dermatol 38:201-206, 1998.
48. Jorgensen MG, Prevalence of amlodipine-related gingival hyperplasia, J Periodontol 68(7):676-8, 1997.
49. van der Vleuten CJ, Trijbels-Smeulders MA, van de Kerkhof PC, Telangiectasia and gingival hyperplasia as side-effects of amlodipine (Norvasc) in a 3-year-old girl, Acta Derm Venereol;79(4):323-4, 1999.
50. Pascual-Castroviejo I, Pascual Pascual SI, Nicardipine-induced gingival hyperplasia, Neurologia 12(1):37-9, 1997.
51. Ikawa K, Ikawa M, Shimauchi H, Iwakura M, Sakamoto S, Treatment of gingival overgrowth induced by manidipine administration. A case report, J Periodontol 73(1):115-22, 2002.
52. Bonnaure-Mallet M, Tricot-Doleux S, Godeau GJ, Changes in extracellular matrix macromolecules in human gingiva after treatment with drugs inducing gingival overgrowth, Arch Oral Biol;40(5):393-400, 1995.
53. Bullon P, Machuca G, Armas JR, Rojas JL, Jimenez G, The gingival inflammatory infiltrate in cardiac patients treated with calcium antagonists, J Clin Periodontol 28(10):897-903, 2001.
54. Nyska A, Shemesh M, Tal H, Dayan D, Gingival hyperplasia induced by calcium-channel blockers: mode of action, Med Hypotheses 43(2):115-8, 1994.
55. Wei JY, Use of calcium entry blockers in elderly patients. Special considerations, Circulation 80(6 Suppl):IV171-7, 1989.
56. Ellis JS, Seymour RA, Steele JG, Robertson P, Butler TJ, Thomason JM, Prevalence of gingival overgrowth induced by calcium-channel blockers: a community-based study, J Periodontol 70(1):63-7, 1999.
57. Monkman SC, Ellis JS, Cholerton S, Thomason JM, Seymour RA, Idle JR, Automated gas chromatographic assay for amlodipine in plasma and gingival crevicular fluid, J Chromatogr B Biomed Appl 678(2):360-4, 1996.
58. Doong H, Dissanayake S, Gowrishankar TR, LaBarbera MC, Lee RC, The 1996 Lindberg Award. Calcium antagonists alter cell shape and induce procollagenase synthesis in keloid and normal human dermal fibroblasts, J Burn Care Rehabil 17(6 Pt 1):497-514, 1996.
59. Coletta RD, Reynolds MA, Martelli-Junior H, Graner E, Almeida OP, Sauk JJ, Testosterone stimulates proliferation and inhibits interleukin-6 production of normal and hereditary gingival fibromatosis fibroblast, Oral Microbiol Immunol 17(3):186-192, 2002.
60. Dayan D, Kozlovsky A, Tal H, Kariv N, Shemesh M, Nyska A, Castration prevents calcium channel blocker-induced gingival hyperplasia in beagle dogs, Hum Exp Toxicol 17(7):396-402, 1998.
61. Border WA, Noble NA, Transforming growth factor beta in tissue fibrosis, N Engl J Med. 331(19):1286-92, 1994.
62. Soory M, Virdi H, Implications of minocycline, platelet-derived growth factor, and transforming growth factor-beta on inflammatory repair potential in the periodontium, J Periodontol 70(10):1136-43, 1999.
63. Sooriyamoorthy M, Gower DB, Eley BM, Androgen metabolism in gingival hyperplasia induced by nifedipine and cyclosporine, J Periodontal Res 25(1):25-30, 1990.
64. Soory M, Kasasa SC, The effects of epidermal growth factor, interleukin-1, and phenytoin, alone and in combination, on C19 steroid conversions in fibroblasts, J Periodontol 68(9):819-26, 1997.
65. Tsele E, Chu AC, Nifedipine and telangiectasias, Lancet 339(8789):365-6, 1992.
66. Silvestre JF, Albares MP, Carnero L, Botella R, Photodistributed felodipine-induced facial telangiectasia, J Am Acad Dermatol 45(2):323-4, 2001.
67. Basarab T, Yu R, Jones RR, Calcium antagonist-induced photo-exposed telangiectasia, Br J Dermatol 136(6):974-5, 1997.
68. Grabczynska SA, Cowley N, Amlodipine induced-photosensitivity presenting as telangiectasia, Br J Dermatol 142(6):1255-6, 2000.
69. Vejlstrup E, Poskitt L, Wojnarowska F, Nifedipine-induced facial telangiectasia, J Eur Acad Dermatol Venereol 5:273-274, 1995.
70. Collins P, Ferguson J, Photodistributed nifedipine-induced facial telangiectasia, Br J Dermatol 129(5):630-3, 1993.
71. Andrisano V, Hrelia P, Gotti R, Leoni A, Cavrini V, Photostability and phototoxicity studies on diltiazem, J Pharm Biomed Anal 25(3-4):589-97, 2001.
72. O'Reilly FM, McKenna D, Murphy GM, Is monochromatic irradiation testing useful in the differentiation of drug-induced photosensitivity from chronic actinic dermatitis? Clin Exp Dermatol, 24(2):118-21, 1999.
73. Karonen T, Stubb S, Keski-Oja J, Truncal telangiectases coinciding with felodipine, Dermatology 196(2):272-3, 1998.
74. Seggev JS, Lagstein Z, Photosensitivity skin reactions to calcium-channel blockers, J Allergy Clin Immunol 97(3):852-5, 1996.
75. Scherschun L, Lee MW, Lim HW, Diltiazem-associated photodistributed hyperpigmentation: a review of 4 cases, Arch Dermatol 137(2):179-82, 2001.
76. Chawla A, Goyal S, Diltiazem-induced hyperpigmentation in an African American woman, J Am Acad Dermatol 46(3):468-9, 2002.
77. Inui S, Itami S, Yoshikawa K, A case of lichenoid purpura possibly caused by diltiazem hydrochloride, J Dermatol 28(2):100-2, 2001.
78. Swale VJ, McGregor JM, Amlodipine-associated lichen planus, Br J Dermatol 144(4):920-1, 2001.
79. Stern R, Khalsa J, Cutaneous adverse reactions associated with calcium-channel blockers, Arch Intern Med 149:829-832, 1989.
80. Cohen AD, Kagen M, Friger M, Halevy S, calcium-channel blockers intake and psoriasis: a case-control study, Acta Derm Venereol 81(5):347-9, 2001.
81. Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH, Calcium regulation of growth and differentiation of mouse epidermal cells in culture, Cell. 19(1):245-54, 1980.
82. Mauro T, Dixon DB, Komuves L, Hanley K, Pappone PA, Keratinocyte K+ channels mediate Ca2+-induced differentiation, J Invest Dermatol 108(6):864-70, 1997.
83. Wakelin SH, James MP, Diltiazem-induced acute generalised exanthematous pustulosis, Clin Exp Dermatol 20(4):341-4, 1995.
84. Vicente-Calleja JM, Aguirre A, Landa N, Crespo V, Gonzalez-Perez R, Diaz-Perez JL, Acute generalized exanthematous pustulosis due to diltiazem: confirmation by patch testing, Br J Dermatol 137(5):837-9, 1997.
85. Roujeau JC, Bioulac-Sage P, Bourseau C, et al. Acute generalized exanthematous pustulosis:Analysis of 63 cases, Arch Dermatol 127:1333-8, 1991.
86. Lavrijsen AP, Van Dijke C, Vermeer BJ, Diltiazem-associated exfoliative dermatitis in a patient with psoriasis, Acta Derm Venereol 66(6):536-8, 1986.
87. Bewley AP, Feher MD, Staughton RC, Erythema multiforme following substitution of amlopidine for nifedipine, BMJ 24;307(6898):241, 1993.
88. Springuel P, Erythema multiforme and nifedipine. CMAJ 1;156(1):90-1, 1997.
89. Lin AY, Baker BA, Verapamil-associated Stevens-Johnson syndrome, DICP 23(12):987-8, 1989.
90. Crowson AN, Magro CM, Subacute cutaneous lupus erythematosus arising in the setting of calcium channel blocker therapy, Hum Pathol 28(1):67-73, 1997.
91. McCauliffe DP, Sontheimer RD, Molecular Characterization of the Ro/SS-A autoantigens, J Invest Dermatol 100(1):73S-79S, 1993.
92. Miyagawa S, Okada N, Inagaki Y, Kitano Y, Ueki H, Sakamoto K, Steinberg ML, SSA/Ro antigen expression in simian virus 40-transformed human keratinocytes, J Invest Dermatol 90(3):342-5, 1988.
93. Brandles LJ, LaBella FS, Histamine and calcium are independently regulated intracellular mediators of lymphocyte mitogenesis, Biochem Biophys Res Commun 31;182(2):786-93, 1992.
94. McMillen MA, Lewis T, Jaffe BM, Wait RB, Verapamil inhibition of lymphocyte proliferation and function in vitro, J Surg Res 39(1),:76-80, 1985.
95. Weir MR, Peppler R, Gomolka D, Handwerger BS, Additive inhibitory effect of cyclosporine and verapamil may occur through different mechanisms that may be dependent or independent of the slow calcium channel, Transplant Proc 21(1 Pt 1):866-70, 1989.
96. Norris DA, Pathomechanisms of photosensitive lupus erythematosus, J Invest Dermatol 100(1):58S-68S, 1993.
97. Norris DA, Lee LA, Antibody-dependent cellular cytotoxicity and skin disease, J Invest Dermatol 85:165S-175S, (suppl 1), 1995.
98. Ameen M, Harman KE, Black MM, Pemphigoid nodularis associated with nifedipine, Br J Dermatol 142(3):575-7, 2000.
99. Bean SF, Good RA, Windhorst DB, Bullous pemphigoid in an 11-year-old boy, Arch Dermatol 102(2):205-8, 1970.
100. Kashihara M, Danno K, Miyachi Y, Horiguchi Y, Imamura S, Bullous pemphigoid-like lesions induced by phenacetin.Report of a case and an immunopathologic study, Arch Dermatol 120(9):1196-9, 1984.
101. Brenner S, Ruocco V, Bialy-Golan A, Tur E, Flaminio C, Ruocco E, Lombardi ML, Pemphigus and pemphigoid-like effects of nifedipine on in vitro cultured normal human skin explants, Int J Dermatol. 38(1):36-40, 1999.
102. Drenth JP, Michiels JJ, Van Joost T, Vuzevski VDVerapamil-induced secondary erythermalgiaBr J Dermatol, 127(3):292-4, 1992.
103. Levesque H, Moore N, Wolfe LM, Courtois H, Erythromelalgia induced by nicardipine (inverse Raynaud's phenomenon?), BMJ 6;298(6682):1252-3, 1989.
104. Cohen JS, Erythromelalgia: new theories and new therapies, J Am Acad Dermatol 43(5 Pt 1):841-7, 2000.
105. Belch JL, Temperature-associated vascular disorders: Raynaud's phenomenon and erythromelalgia, In: Lowe GD, Tooke JE, editors. A textbook of vascular medicine. London University Press; p.339-52, 1996.
106. Mork C, Kvernebo K, Asker CL, Salerud EG, Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism, J Invest Dermatol. 119(4):949-53, 2002.
107. Braverman IM, The cutaneous microcirculation, J Investig Dermatol Symp Proc 5(1):3-9, 2000.
108. Otto C, Richter WO, Unilateral gynecomastia induced by treatment with diltiazem, Arch Intern Med 14;154(3):351, 1994.
109. Braunstein GD, Gynecomastia, N Engl J Med 18;328(7):490-5, 1993.
110. Zochling J, Large G, Fassett R, Gynaecomastia and amlodipine, Med J Aust 20;160(12):807, 1994.
111. Romeo JH, Dombrowski R, Kwak YS, Fuehrer S, Aron DC, Hyperprolactinaemia and verapamil: prevalence and potential association with hypogonadism in men, Clin Endocrinol (Oxf), 45(5):571-5, 1996.
112. Ortega Calvo M, Macias Perez V, Merino Kolly MN, Torello Iserte J. Hyperprolactinemia secondary to hypotensive treatment with verapamil, Aten Primaria. 26(7):510-1, 2000.
113. Kelley SR, Kamal TJ, Molitch ME, Mechanism of verapamil calcium channel blockade-induced hyperprolactinemia, Am J Physiol. 270(1 Pt 1):E96-100, 1996.
114. Barbaro D, Faggionato F, Pallini S, Carnesecchi C, Palla A, Bombara M, Falciani C, La Gioia A, Loni G, Verapamil acute administration: a new dynamic test in hyperprolactinemic states, Metabolism. 48(11):1351-6, 1999.
115. Veldhuis JD, Borges JL, Drake CR, Rogol AD, Kaiser DL, Thorner MO, Divergent influences of the structurally dissimilar calcium entry blockers, diltiazem and verapamil, on thyrotropin- and gonadotropin-releasing hormone-stimulated anterior pituitary hormone secretion in man. J Clin Endocrinol Metab. 60(1):144-9, 1985.
116. Satoh T, Fujita KI, Munakata H, Itoh S, Nakamura K, Kamataki T, Itoh S, Yoshizawa I, Studies on the interactions between drugs and estrogen: analytical method for prediction system of gynecomastia induced by drugs on the inhibitory metabolism of estradiol using Escherichia coli coexpressing human CYP3A4 with human NADPH-cytochrome P450 reductase, Anal Biochem 15;286(2):179-86, 2000.
117. Galbraith RA, Michnovicz JJ, The effects of cimetidine on the oxidative metabolism of estradiol, N Engl J Med. 321(5):269-74, 1989.
118. Causes of hyperprolactinemia, UpToDate(r), 2002.
119. Cohen DM, Bhattacharyya I, Lydiatt WM, Recalcitrant oral ulcers caused by calcium-channel blockers: diagnosis and treatment considerations. J Am Dent Assoc 130(11):1611-8, 1999 .
120. Malhotra HS, Plosker GL, Barnidipine. Drugs 61(7):989-96; discussion 997-8, 2001.
121. Lim AC, Hart K, Murrell D, A granuloma annulare-like eruption associated with the use of amlodipine. Australas J Dermatol 43(1):24-7, 2002.
122. Bosch X, Campistol JM, Botey A, Cases A, Revert L, Nifedipine-induced parotitis, Lancet 2(8504):467, 1986.
123. Massimo C, Sebastianelli G, Noera G. Nifedipine-induced parotitis: a hypersensitivity reaction, Am J Cardiol. 61(10):874, 1988.
124. Bosch X, Sobrino J, Lopez-Soto A, Urbano-Marquez A, Parotitis due to nicardipine, BMJ. 4;304(6831):882, 1992.
125. Odeh M, Oliven A, Verapamil-associated liver injury, Harefuah 134(1):36-7, 1998.
126. Read GM, Verapamil and hair colour change, Lancet 14;338(8781):1520, 1991.
127. Wirebaugh SR, Geraets DR, Reports of erythematous macular skin eruptions associated with diltiazem therapy. DICP 24(11):1046-9, 1990.
128. Luscher TF, Waeber B, Calcium antagonists as first-line therapy in hypertension: results of the Swiss Isradipine Study. Swiss Hypertension Society. J Cardiovasc Pharmacol 18 Suppl 3:S1-3, 1991.
129. del Rio Fernandez MC, Plagaro Cordero ME, de Frutos Arribas JF, del Pozo Roman T, Martin Escudero JC, Leukocytoclastic vasculitis in relation to amlodipine administration, Rev Clin Esp 195(10):738-9, 1995.
.
© 2003 Dermatology Online Journal