Papillon-Lefèvre syndrome: Case report and review of the literature
Published Web Location
https://doi.org/10.5070/D39419g7svMain Content
Papillon-Lefèvre syndrome: Case report and review of the literature
Shahbaz A Janjua MD1, Amor Khachemoune MD2
Dermatology Online Journal 10 (1): 13
From the Ayza Skin and Research Center,Lalamusa, Pakistan1, and the Division of Dermatology, Georgetown University Medical Center, Washington DC2. amorkh@pol.net
Abstract
A 15-year-old boy presented with symmetric, well-demarcated, yellowish, keratotic plaques over the skin of his palms and soles extending onto the dorsal surfaces. Well-circumscribed, psoriasiform, erythematous, scaly plaques were also present on the elbows and knees bilaterally along with dystrophy and transverse grooving of the nails. He also had swollen and friable gums since the age of 3 with subsequent loss of most of his permanent dentition. These findings are consistent with Papillon-Lefèvre syndrome. The clinical presentation, differential diagnosis, complications and management of this syndrome are discussed.
Clinical summary

|
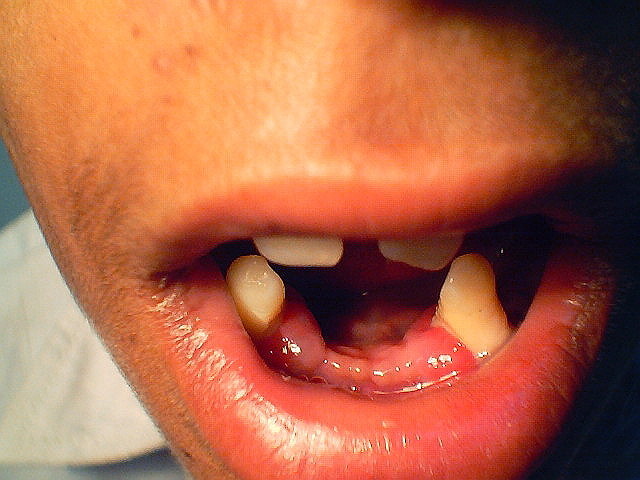
|
| Figure 1 | Figure 2 |
|---|
History.—A 15-year-old boy presented with persistent thickening, flaking and scaling of the skin of his palms and soles associated with recurrently swollen and friable gums since age of 3. He also had of premature shedding of his deciduous teeth and loss of most of his permanent teeth. The remainder of his past medical history was unremarkable. There was no family history of ichthyosis or hereditary or acquired palmoplantar keratodermas.
Physical examination.— There were symmetric, well-demarcated, yellowish, keratotic, confluent plaques affecting the skin of his palms and soles and extending onto the dorsal surfaces. His nails showed dystrophy and transverse grooving. Well-circumscribed, psoriasiform, erythematous, scaly plaques were present on the elbows and knees bilaterally. On oral examination, his gums were edematous, friable and receding with loss of most teeth except the upper central incisors, lower canines and upper and lower molars. (Figs. 1 and 2).
Laboratory data.—Complete blood count, liver function transaminase levels, total bilirubin, and alkaline phosphatase were normal.
Histopathology.—None. The parents declined a biopsy.
Diagnosis..—Papillon-Lefèvre syndrome (PLS)
Comment
Papillon-Lefèvre syndrome, first described by two French physicians Papillon and Lefèvere in 1924 [1], is an extremely rare genodermatosis inherited as an autosomal recessive trait, affecting children between the ages 1-4 years [2, 3]. It has a prevalence of 1-4 cases per million persons [3]. Males and females are equally affected and there is no racial predominance [4]. The disorder is characterized by diffuse palmoplantar keratoderma and premature loss of both deciduous and permanent teeth. The palmoplantar keratoderma typically has its onset between the ages 1 and 4 years [5]. The sharply demarcated erythematous keratotic plaques may occur focally, but usually involve the entire surface of the palms and soles, sometimes extending onto the dorsal surfaces of the hands and feet [6]. Often, there is associated hyperhidrosis of the palms and soles resulting in a foul-smelling odor. [3] Well-demarcated psoriasiform plaques occur on the elbows and knees [6]. The findings may worsen in winter and be associated with painful fissures.
The second major feature of PLS is severe periodontitis, which starts at age 3 or 4 years [6]. The development and eruption of the deciduous teeth proceed normally, but their eruption is associated with gingival inflammation and subsequent rapid destruction of the periodontium. The resulting periodontitis characteristically is unresponsive to traditional periodontal treatment modalities and the primary dentition is usually exfoliated prematurely by age 4 years. After exfoliation, the inflammation subsides and the gingiva appears healthy. However, with the eruption of the permanent dentition the process of gingivitis and periodontitis is usually repeated and there is subsequent premature exfoliation of the permanent teeth, although the third molars are sometimes spared [7, 8]. The degree of dermatologic involvement may not be related to the level of periodontal infection [9]. Nail changes are apparent in advanced cases as in this case, manifested by transverse grooving and fissuring [10]. In addition to the skin and oral findings, patients may have decreased neutrophil, lymphocyte, or monocyte functions and an increased susceptibility to bacteria, associated with recurrent pyogenic infections of the skin [11]. Pyogenic liver abscess is increasingly recognized as a complication of PLS associated with impairment of the immune system [11].
Another feature of PLS may be radiographic evidence of intracranial calcification [12].
Histopathological findings of affected skin have not been well described in the literature. Reported findings have consisted of hyperkeratosis, occasional patches of parakeratosis, acanthosis, and a slight perivascular inflammatory infiltrate. [13]
The cause of PLS is not well understood, but recently, 2 research groups have reported that loss-of-function mutations affecting both the alleles of the cathepsin-C gene, located on chromosome 11q14.1-q14.3, were associated with PLS [7, 14]. The cathepsin C gene encodes a cysteine-lysosomal protease also known as dipeptidyl-peptidase I, which functions to remove dipeptides from the amino terminus of the protein substrate. It also has endopeptidase activity. The cathepsin-C gene is expressed in epithelial regions commonly affected by PLS such as palms, soles, knees, and keratinized oral gingiva. It is also expressed at high levels in various immune cells including polymorphonuclear leukocytes, macrophages, and their precursors. Several mutations have been reported in the cathepsin-C gene in individuals from diverse ethnic groups [13, 14]. An interesting feature of the cathepsin C gene is that mutations in this gene also result in two other closely related conditions: the Haim-Munk syndrome, and prepubertal periodontitis. A common clinical manifestation in all three syndromes is severe early-onset periodontitis [18]. An increased prevalence of parental consanguinity has been reported in PLS patients [3]. All PLS patients are homozygous for the same cathepsin-C mutations inherited from a common ancestor. Parents and siblings, heterozygous for cathepsin C mutations do not show either the palmoplantar hyperkeratosis or severe early onset periodontitis characteristic of PLS [7]. Thus Haim-Munk syndrome, prepubertal periondontitis and PLS seem to be allelic variants. The exact cause of the periodontal disease in PLS has not been found but it has been attributed to decreased neutrophil phagocytosis, bacterial infection and impaired reactivity to T- and B-cell mitogens. The exact mechanism of the increased susceptibility to infections is also unknown, but some investigators have demonstrated a dysfunction in neutrophil motility and bactericidal function [15, 16, 17, 18]. It would be pertinent to mention that there are reports of at least 6 cases of late-onset variation of PLS without underlying cathepsin-C gene mutations [19].
The palmoplantar keratodermas (PPKs) are a heterogeneous group of keratinization disorders characterized by erythema and hyperkeratosis of the palms and soles. More than 40 different types of PPK have been described [15]. The reclassification of the primary PPKs (diffuse, focal, punctate, and palmoplantar ectodermal dysplasias) proposed by Stevens et al. [16] distinguished those conditions in which the cutaneous abnormalities are restricted to the epidermis (PPK types I, II, and III) from those conditions that presented with a wider ectodermal abnormality (PPK type IV). All of those conditions in which cutaneous lesions are restricted to the epidermis (types I, II, and III) are either acquired or are transmitted as autosomal dominant traits.
PLS is an uncommon autosomal recessive type-IV palmoplantar ectodermal dysplasia. Of the palmoplantar ectodermal dysplasias, only PLS and Haim-Munk syndrome (HMS) are associated with premature periodontal destruction.
As mentioned earlier, PLS, HMS and prepubertal periodontitis have been described as allelic variants; we will consider only HMS and premature periodontitis in the differential diagnosis of PLS.
Haim Munk syndrome has been described as an autosomal-recessive genodermatosis characterized by congenital palmoplantar keratoderma and progressive early-onset perodontitis. It was first reported in 1965 by Haim and Munk in Jewish families from Cochin, India on the Malabar Coast [17]. Hart et al. identified a mutation of cathepsin C affecting a highly conserved amino-acid residue in Haim-Munk syndrome, demonstrating that PLS and Haim-Munk syndromes are allelic disorders [18]. In addition to palmoplantar keratosis and periodontitis, other clinical findings include recurrent pyogenic skin infections, acro-osteolysis, atrophic changes of the nails, arachnodactyly, and a peculiar radiographic deformity of the fingers consisting of tapered, pointed phalangeal ends, claw-like volar curve, and pes planus. In contrast to PLS, the cutaneous findings in HMS have been reported to be more severe and extensive. The periodontium in HMS may be less severely affected than in PLS, but gingival inflammation and alveolar-bone destruction are present and severe. Although the palmoplantar findings and severe periodontitis are suggestive of PLS, the association of other clinical features, particularly nail deformities and arachnodactyly are suggestive of HMS being a distinct disorder [18].
Prepubertal periodontitis is another rare genodermatosis with an etiology attributed to a cathepsin-C gene mutation. It is characterized by rapidly progressive early-onset periodontitis with destruction of the periodontium of the deciduous and permanent teeth. Prepubertal periodontitis may be localized or generalized. It may occur as part of a recognized syndrome or as an isolated non-syndromic disorder. Both autosomal dominant and autosomal recessive patterns of familial transmission have been described for prepubertal periodontitis. Identification of a cathepsin-C missense mutation indicates that this form of non-syndromic prepubertal periodontitis is also an allelic variant of type-IV palmoplantar ectodermal dysplasia. The radiographic presentation of alveolar bone loss in prepubertal periodontitis in many cases appears similar to that observed in PLS, but it is differentiated from PLS by the absence of associated palmoplantar keratoderma [18].
A multidisciplinary approach is important for the care of patients with PLS. The skin manifestations of PPK are usually treated with emollients [6]. Salicylic acid and urea may be added to enhance their effects [11]. Oral retinoids including acitretin, etretinate, and isotretinoin are the mainstay of the treatment of both the keratoderma and periodontitis associated with PLS. Treatment may be more beneficial if it is started during the eruption and maintained during the development of the permanent teeth [20, 21]. The periodontitis in PLS is usually difficult to control. Effective treatment for the periodontitis includes extraction of the primary teeth combined with oral antibiotics and professional teeth cleaning [11, 22]. It is reported that etretinate and acitretin modulate the course of periodontitis and preserve the teeth [23]. A course of antibiotics should be tried to control the active periodontitis in an effort to preserve the teeth and to prevent bacteremia and subsequently pyogenic liver abscess. [11]. The risk of pyogenic liver abscess should be kept in mind in evaluating these patients when they present with fever of unknown origin [11].
References
1. Papillon MM, Lefèvre P. Deux cas de keratodermie palmaire et plantaire symmetrique famiale (maladie de Meleda) chez le frère et la soeur: coéxistance dans les deux cas d'altérations dentaires graves. Bull Soc Derma Symp 1924;31:82-7.2. Hart TC, Shapira L. Papillon-Lefèvre syndrome. Periodontol 2000. 1994 Oct;6:88-100.
3. Gorlin RJ, Sedano H, Anderson VE. The syndrome of palmar-plantar hyperkeratosis and premature periodontal destruction of the teeth. J Pediatr 1964;65:895-908.
4: Cury VF, Costa JE, Gomez RS, Boson WL, Loures CG, De ML. A novel mutation of the cathepsin C gene in Papillon-Lefèvre syndrome. J Periodontol. 2002 Mar;73(3):307-12.
5. Bach JN, Levan NE. Papillon-Lefèvre syndrome. Arch Dermatol. 1968 Feb;97(2):154-8.
6. Siragusa M, Romano C, Batticane N, Batolo D, Schepis C. A new family with Papillon-Lefèvre syndrome: effectiveness of etretinate treatment. Cutis. 2000 Mar;65(3):151-5.
7. Hart TC, Hart PS, Bowden DW, Michalec MD, Callison SA, Walker SJ, Zhang Y, Firatli E. Mutations of the cathepsin C gene are responsible for Papillon-Lefèvre syndrome. J Med Genet. 1999 Dec;36(12):881-7.
8. Haneke E. The Papillon-Lefèvre syndrome: keratosis palmoplantaris with periodontopathy. Report of a case and review of the cases in the literature. Hum Genet. 1979 Sep 2;51(1):1-35.
9. Ullbro C, Crossner CG, Nederfors T, Alfadley A, Thestrup-Pedersen K. Dermatologic and oral findings in a cohort of 47 patients with Papillon-Lefèvre syndrome. J Am Acad Dermatol. 2003 Mar;48(3):345-51.
10. Giansanti JS, Hrabak RP, Waldron CA.Palmar-plantar hyperkeratosis and concomitant periodontal destruction (papillon-Lefèvre syndrome). Oral Surg Oral Med Oral Pathol. 1973 Jul;36(1):40-8.
11. Almuneef M, Al Khenaizan S, Al Ajaji S, Al-Anazi A. Pyogenic liver abscess and Papillon-Lefèvre syndrome: not a rare association. Pediatrics. 2003 Jan;111(1):e85-8.
12. Reyes VO, King-Ismael D, Abad-Venida L. Papillon-Lefèvre syndrome. Int J Dermatol. 1998 Apr;37(4):268-70.
13. Angel TA, Hsu S, Kornbleuth SI, Kornbleuth J, Kramer EM. Papillon-Lefèvre syndrome: a case report of four affected siblings. J Am Acad Dermatol. 2002 Feb;46(2 Suppl Case Reports):S8-10.
14. Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG, Markham A, Wong M, Widmer R, Ghaffar KA, Pemberton M, Hussein IR, Temtamy SA, Davies R, Read AP, Sloan P, Dixon MJ, Thakker NS. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet. 1999 Dec;23(4):421-4.
15. Itin PH. Classification of autosomal dominant palmoplantar keratoderma: past-present-future. Dermatology 1992;185:163-165.
16. Stevens HP, Kelsell DP, Bryant SP, Bishop DT, Spurr NK, Weissenbach J, Marger D, Marger RS, Leigh IM. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Literature survey and proposed updated classification of the keratodermas. Arch Dermatol 1996;132:640-651.
17. Haim S, Munk J. Keratosis palmo-plantaris congenita, with periodontosis, arachnodactyly, and peculiar deformity of the terminal phalanges. Br J Dermatol 1965;77:42-54.
18. Hart TC, Hart PS, Michalec MD, Zhang Y, Firatli E, Van Dyke TE, Stabholz A, Zlotogorski A, Shapira L, Soskolne WA. Haim-Munk syndrome and Papillon-Lefèvre syndrome are allelic mutations in cathepsin C. J Med Genet. 2000 Feb;37(2):88-94. Erratum in: J Med Genet 2001 Jan;38(1):79.
19. Pilger U, Hennies HC, Truschnegg A, Aberer E. Late-onset Papillon-Lefevre syndrome without alteration of the cathepsin C gene. J Am Acad Dermatol. 2003 Nov;49(5 Suppl):240-3.
20. Al-Khenaizan S. Papillon-Lefèvre syndrome: the response to acitretin. Int J Dermatol. 2002 Dec;41(12):938-41.
21. Blanchet-Bardon C, Nazzaro V, Rognin C, Geiger JM, Puissant A. Acitretin in the treatment of severe disorders of keratinization. Results of an open study. J Am Acad Dermatol. 1991 Jun;24(6 Pt 1):982-6.
22. Ishikawa I, Umeda M, Laosrisin N. Clinical, bacteriological, and immunological examinations and the treatment process of two Papillon-Lefèvre syndrome patients. J Periodontol. 1994 Apr;65(4):364-71.
23. Galmetti C, Nazzaro V, Cerri D, Fracasso L. Long-term preservation of permanent teeth in a patient with Papillon-Lefèvre syndrome treated with etretinate. Pediatr Dermatol. 1989 Sep;6(3):222-5.
© 2004 Dermatology Online Journal