Epidermolysis bullosa simplex with mottled pigmentation
Published Web Location
https://doi.org/10.5070/D38sr2q5d7Main Content
Epidermolysis bullosa simplex with mottled pigmentation
John C Browning MD1,2, Brooke Mohr BA1
Dermatology Online Journal 18 (1): 9
1. Division of Dermatology and Cutaneous Surgery2. Department of Pediatrics, Pediatric Dermatology Section
The University of Texas Health Science Center at San Antonio, San Antonio, Texas
Abstract
Epidermolysis bullosa is a rare disorder with several variants. Included in this disorder is epidermolysis bullosa with mottled pigmentation (EBS-MP). We report a case of a young child with this rare disorder and explain the genetic cause.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|
A 7-year-old male presented with a history of “blisters and bumps” on his hands and feet that first appeared when he was 6 months old. They seemed to appear with heavy activity or trauma. The child denies burning or itching but complains of pain with the blisters at times. Around the same time that the blisters appeared, the child also developed mottled pigmentation on his trunk and extremities. There was no history of blisters or trauma to the skin prior to development of the pigmentation. Past medical history is unremarkable. There is no family history of blistering or pigmentary anomalies.
 |
| Figure 3 |
|---|
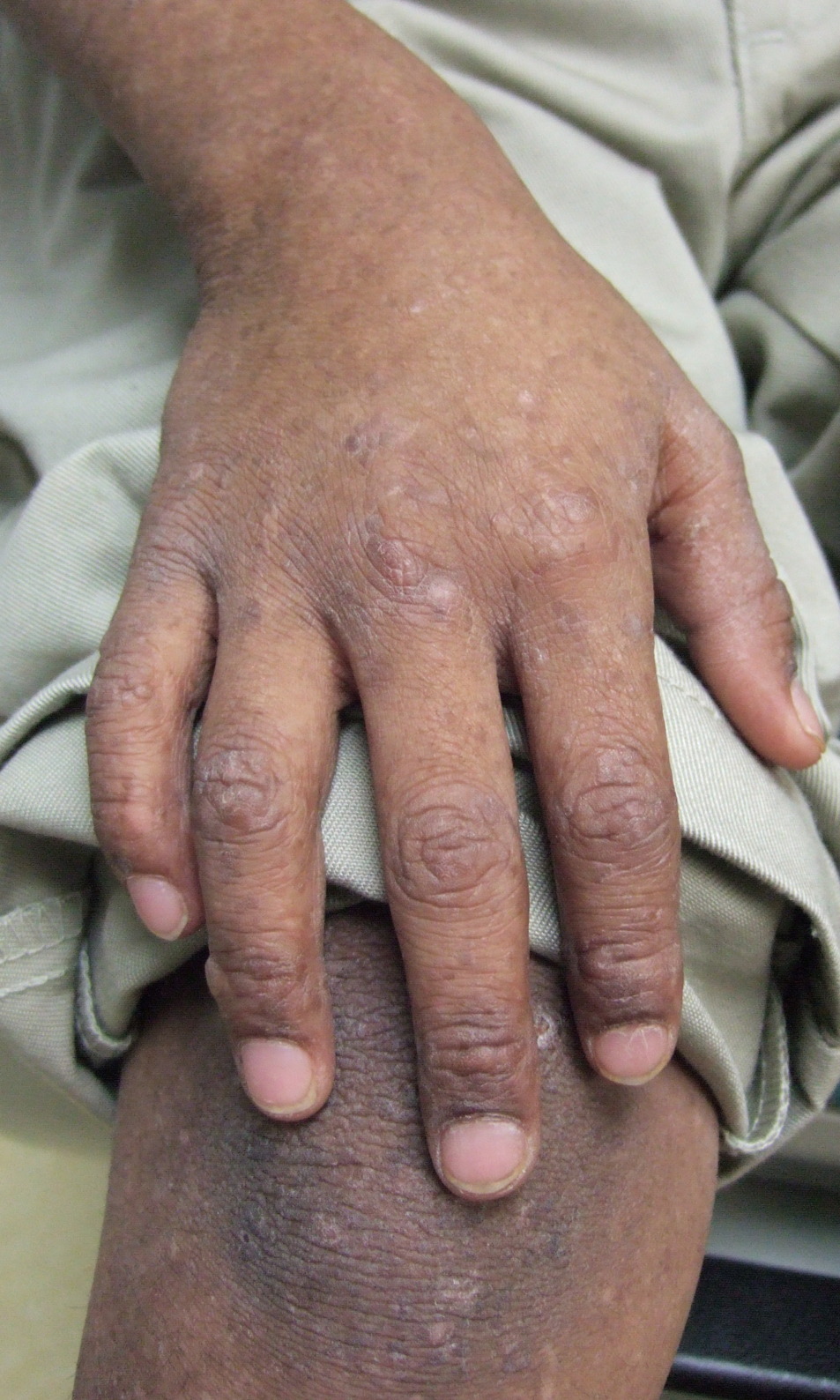
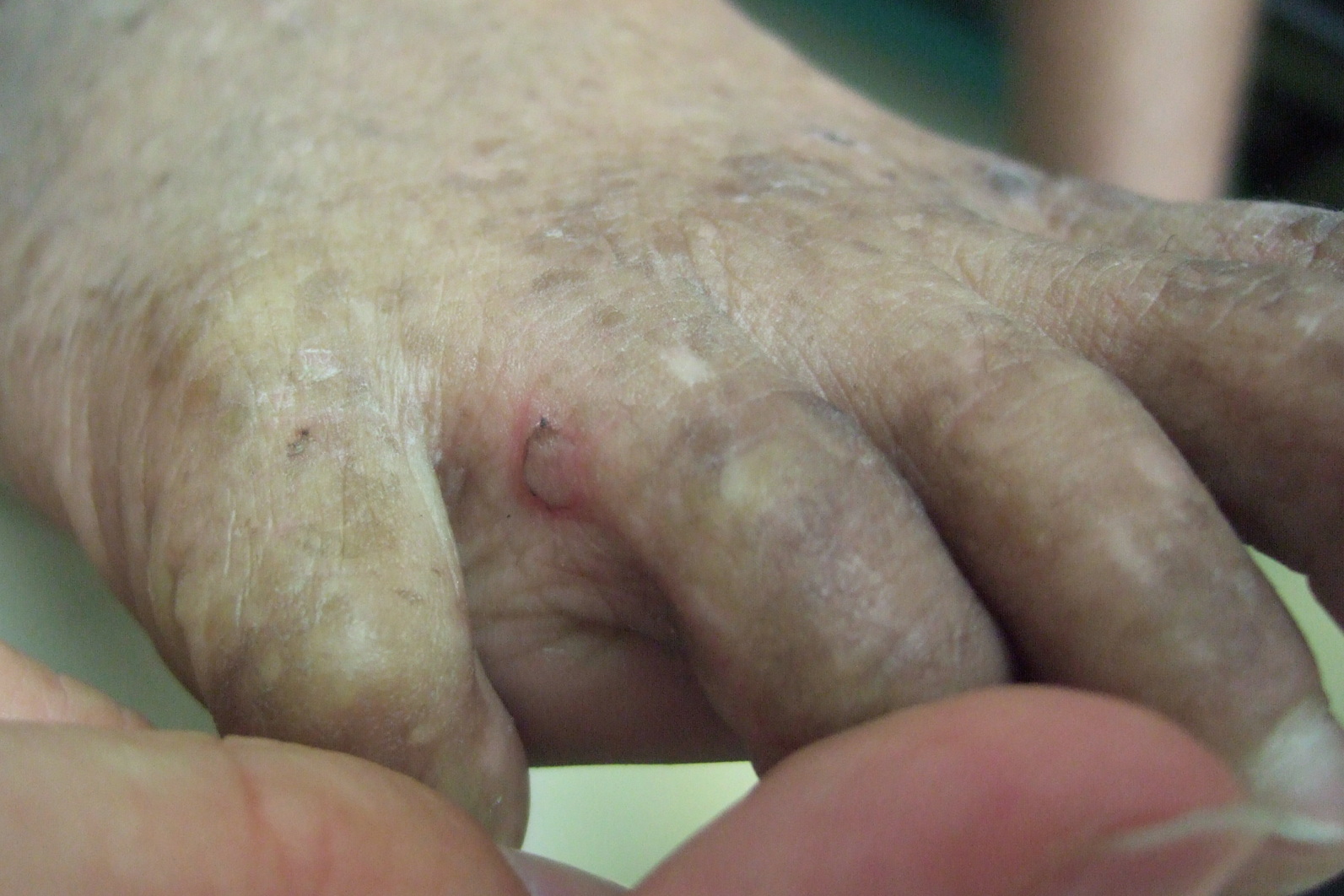
On physical exam the child is active and playful. He has mottled brown hyperpigmentation involving the axillae, dorsal forearms, anterior shins, and dorsal feet (Figure 1). He also has verrucous papules over the knees, elbows, wrists, and feet (Figure 2). There is a small blister between the 4th and 5th digits on the left foot (Figure 3). Nails, palms, and soles are normal.
A diagnosis of epidermolysis bullosa simplex with mottled pigmentation (EBS-MP) was suspected. A skin biopsy to be sent for direct immunofluorescent mapping was offered but was refused by the mother and child because of fear of pain with the procedure. Instead genetic testing was performed and the child was found to be heterozygous for the P25L mutation in the KRT5 gene, confirming a diagnosis of EBS-MP. Specifically, the child was found to be heterozygous for a C>T nucleotide substitution in exon 1, resulting in the replacement of a proline codon with a leucine codon at amino acid position 25 in the KRT5 gene. The P25L missense mutation in the KRT5 gene has been reported in the majority of patients with EBS-MP. Because there was no family history of EBS-MP, we believe this to be a new mutation. Genetic counseling was provided regarding the risk of EBS-MP in future offspring of the child. Specifically, each offspring has a 50 percent chance of having EBS-MP.
Discussion
EBS-MP is an autosomal dominant disorder that can be sporadic in the absence of a positive family history. The disease is characterized by intraepidermal blisters that occur with minimal trauma, mottled pigmentation on the limbs and trunk, nail dystrophy (lacking in our patient), and hyperkeratotic papules over extensor surfaces. The hyper- and hypopigmentation usually begins in infancy, spreading slowly from the extremities to the trunk [1].
The diagnosis can be suspected clinically, especially with a positive family history. It can be confirmed by skin biopsy of an induced blister with immunofluorescent (IF) antigen mapping or electronmicroscopy or by genetic testing. Routine histology is not helpful. The gold standard for diagnosis is genetic testing because at least one case of EBS-MP has been reported in the literature with discordant results between genetic testing and IF mapping [1].
Biopsy of a hyperpigmented area will show focal hyperpigmentation of the basal cells and pigment incontinence without an inflammatory infiltrate [2].
EBS-MP is most often related to the P25L missense mutation in the KRT5 gene but it has also been reported from mutations in KRT14 [3, 4]. It is unknown why pigmentary alterations occur in EBS- MP and not other subtypes of epidermolysis bullosa. It is interesting that Dowling-Degos disease, also known as pigmented reticulate anomaly of the flexures, is also caused by KRT5 mutations and is characterized by progressive reticulate hyperpigmentation of the flexures [1].
Treatment of EBS-MP is focused on reducing formation of blisters. This can be achieved by using loose-fitting or padded shoes. Bullae, when formed, can be decompressed using an 18-gauge needle. There is no treatment for the hyperpigmentation. Because EBS-MP is an autosomal dominant disorder, it is important for affected individuals to understand that there is a 50 percent possibility of having an affected child with each pregnancy.
References
1. Andres C, Chen W, Hofmann H, Ring J, Schnopp C. Epidermolysis bullosa simplex with mottled pigmentation: a case report. Int J Dermatol. 2009 Jul;48(7):753-4. [PubMed]2. Bruckner-Tuderman L, Vogel A, Rüegger S, Odermatt B, Tönz O, Schnyder UW. Epidermolysis bullosa simplex with mottled pigmentation. J Am Acad Dermatol. 1989 Aug;21(2 Pt 2):425-32. [PubMed]
3. Harel A, Bergman R, Indelman M, Sprecher E. Epidermolysis bullosa simplex with mottled pigmentation resulting from a recurrent mutation in KRT14. J Invest Dermatol. 2006 Jul;126(7):1654-7. Epub 2006 Apr 6. [PubMed]
4. Gray C, Greenlaw SM, Alavian C, Wiss K. Epidermolysis Bullosa Simplex With Mottled Pigmentation: A Novel KRT14 Mutation. J Drugs Dermatol. 2011 Aug 1;10(8):926-7. [PubMed]
© 2012 Dermatology Online Journal