Piebaldism and neurofibromatosis type 1: family report
Published Web Location
https://doi.org/10.5070/D38pg2d4szMain Content
Piebaldism and neurofibromatosis type 1: family report
Ana Filipa Duarte1,2, Alberto Mota1,2, Teresa Baudrier1, Paulo Morais1,2, António Santos1, Rita Cerqueira3, Purificação Tavares3, Filomena Azevedo1
Dermatology Online Journal 16 (1): 11
1. Department of Dermatology, Hospital de São João, EPE, Porto, Portugal. duarte.af.t30@gmail.com2. Faculty of Medicine, Porto, Portugal
3. CGC Centro de Genética Clínica, Porto, Portugal
Abstract
Piebaldism is a rare disorder present at birth and inherited as an autosomal dominant trait. It results from a mutation in the c-kit proto-oncogene and is associated with a defect in the migration and differentiation of melanoblasts from the neural crest. Clinical manifestations and phenotypic severity strongly correlates with the site of mutation within the KIT gene. Here we report a 3-year-old boy and his 33-year-old father with leukoderma and poliosis associated with clinical criteria for Neurofibromatosis type 1. Genetic study of both revealed a p.Gly610Asp mutation in the KIT gene. This familiar mutation has not yet been reported in the literature. There are rare reports of piebaldism in association with neurofibromatosis type I.
Introduction
Piebaldism is a rare disorder, characterized by congenital localized leukoderma and leukotrichia, affecting all races with equal gender distribution [1]. It is inherited as an autosomal dominant trait and results from a mutation in the c-kit proto-oncogene, mapped to the proximal long arm of chromosome 4 (4q12), which encodes a cell-surface tyrosine-kinase receptor whose ligands are a stem cell and mast cell growth factor. C-kit is expressed in a large variety of cells, including melanocytes and mast cells; it has a role in melanogenesis. The mutation of this receptor is associated with a defect in the migration and differentiation of melanoblasts from the neural crest. It seems also to be important for hemtopoiesis and gametogenesis [2, 3]. Several pathologic mutations of the c-kit proto-oncogene have been identified in 75 percent of patients with piebaldism [4]. Clinical manifestations and phenotypic severity strongly correlates with the mutation site within the KIT gene [1]. In fact, dominant negative missense mutations of the intracellular tyrosine kinase domain appear to yield the most severe phenotypes. Intermediate phenotypes are seen with mutations near the transmembrane region, whereas mutations in the aminoterminal extracellular ligand binding domain are associated with the mildest forms of piebaldism [1, 5]. Recently, deletions in the SLUG gene, a zinc-finger neural crest transcription factor, have been reported in patients with piebaldism that lacked mutations in KIT [6]. It is also interesting to note that cutaneous graft-versus-host disease has been reported as affecting only areas of piebaldism, suggesting that piebaldism-affected skin may be immunologically different from unaffected skin [7].
Piebaldism is characterized by the absence of melanocytes in affected areas of the skin and hair. It presents at birth with bilateral depigmentated and irregularly shaped patches, with typical hyperpigmentation or normal pigmentation islands within and at the borders of the depigmented areas [1]. A white forelock is present in 80-90 percent of the patients [1, 2].
Neurofibromatosis type 1 (NF1) is an autosomal dominant neurocutaneous disorder. The NF1 gene belongs to the family of tumor suppressor genes and encodes a widely expressed protein called neurofibromin, which is a major negative regulator of the Ras oncogene pathway, participating in the control of cell growth and differentiation [8]. Despite progress in understanding the molecular basis, the diagnosis of NF1 is based on clinical criteria established by the National Institute of Health (NIH) Consensus Conference in 1987 [9]. According to these criteria, two or more of the following findings establish the diagnosis of NF1: a) six or more café-au-lait spots greater than 5 mm in diameter in prepubertal individuals and more than 15 mm after puberty, b) two or more neurofibromas or one plexiform neurofibroma, c) freckling in the axilary or inguinal regions, d) optic pathway tumor, e) two or more Lisch nodules (iris hamartomas), f) distinctive bone lesions such as sphenoid wing dysplasia and g) a first-degree relative with NF1 [2, 9, 10].
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
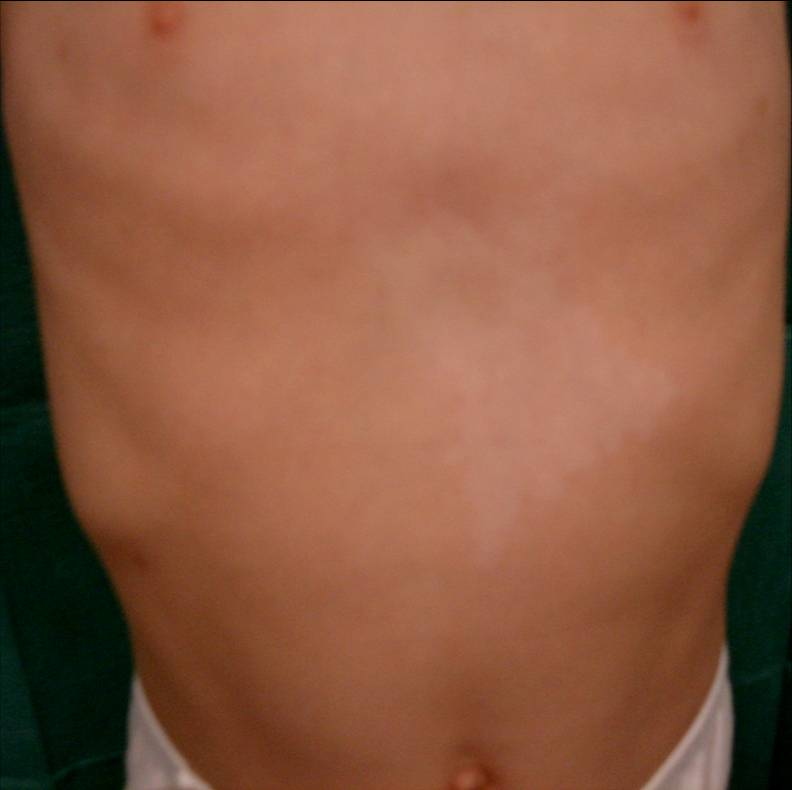
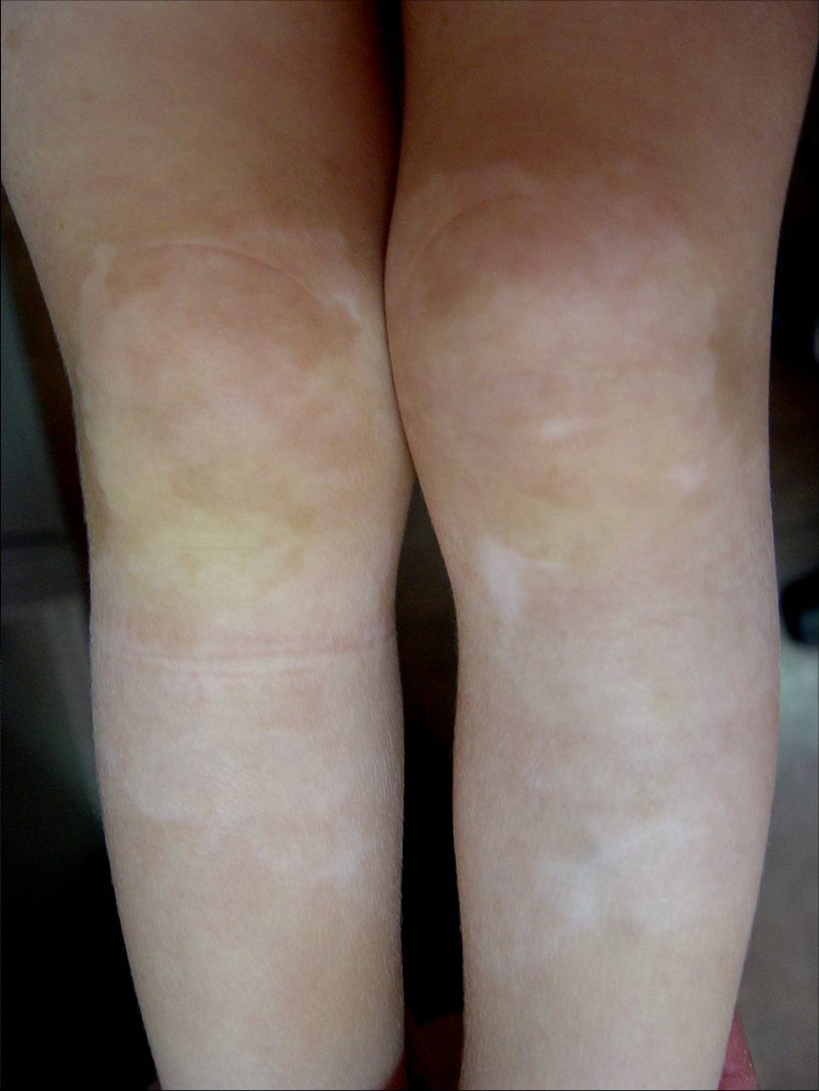
| Figure 1. Patches of hypopigmentation and depigmentation on the chest with islands of normal pigmentation within them Figure 2. Symmetrical patches of hypopigmentation and depigmentation on the lower extremities, with islands of normal pigmentation within them | |
 |
| Figure 3 |
|---|
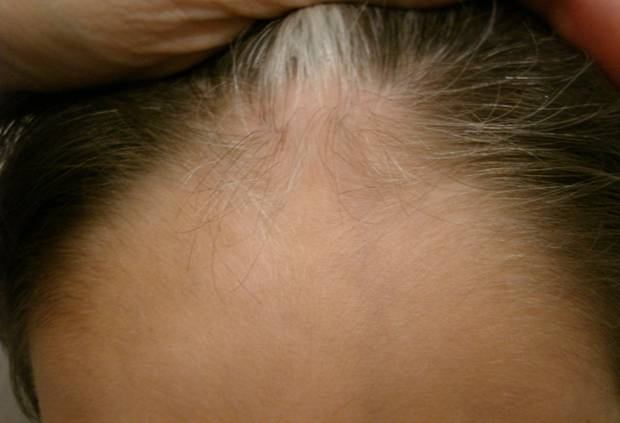
| Figure 3. White forelock |
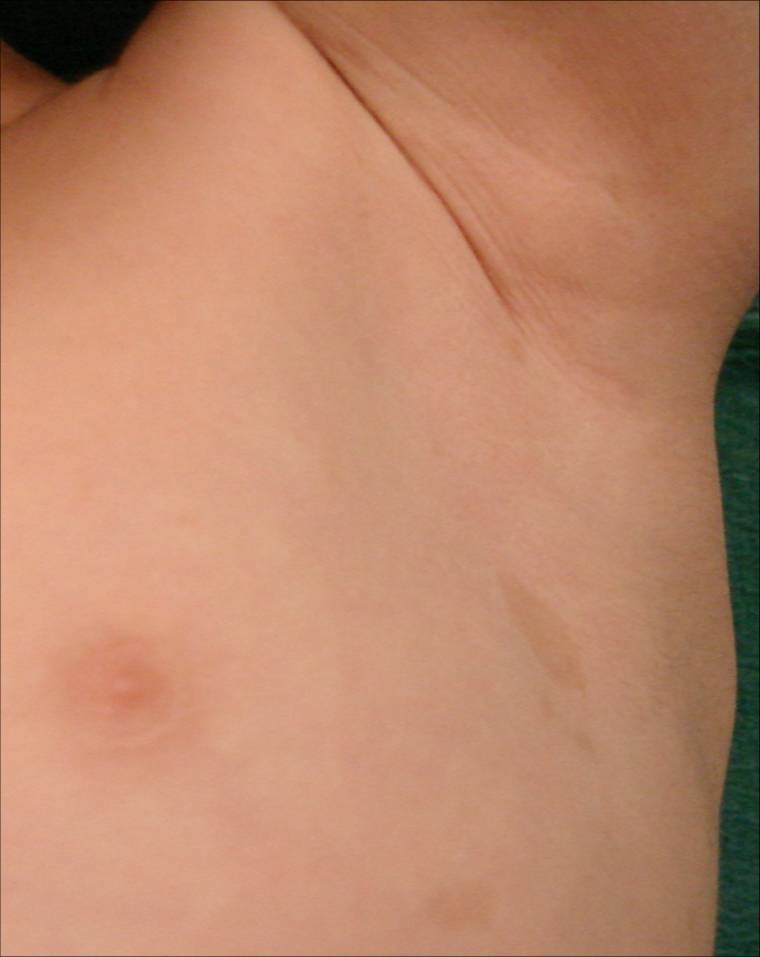
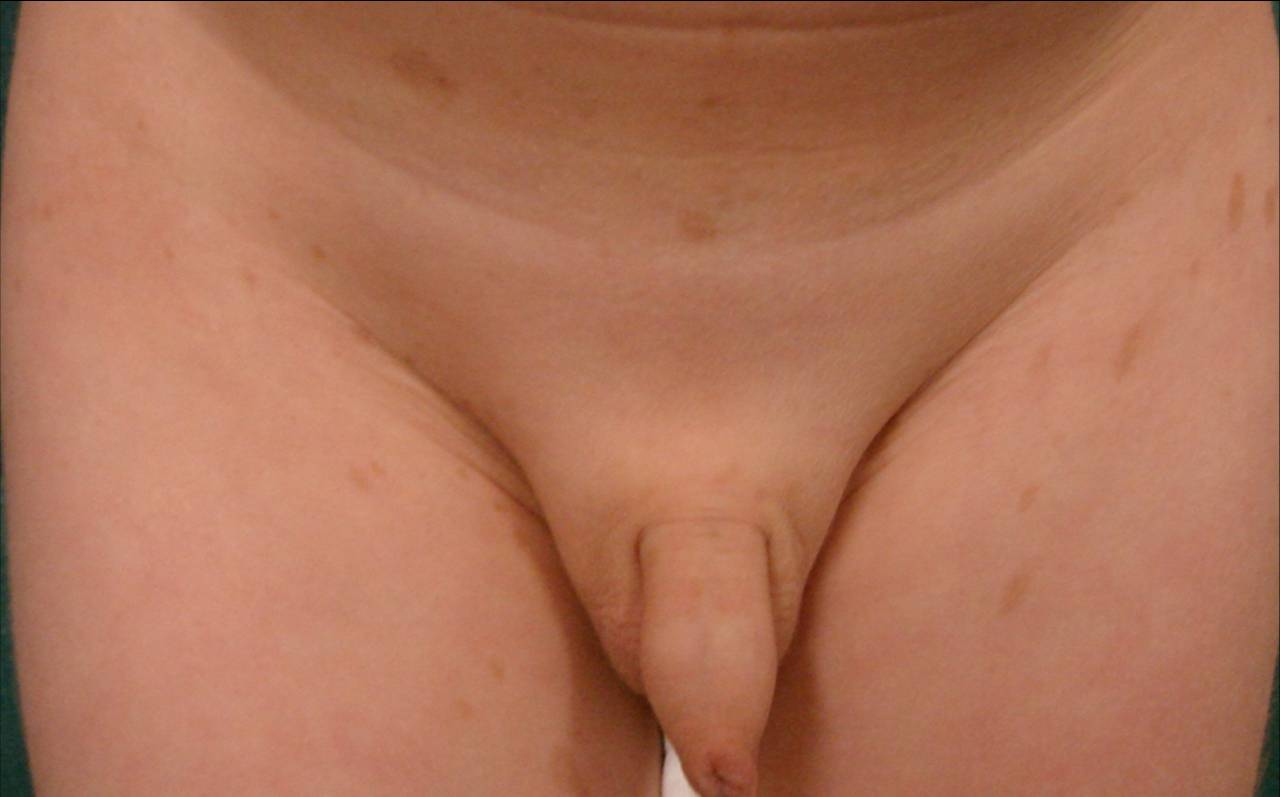
Here we report the case of a 3-year-old boy brought to our Dermatology department because of a pigment disorder since birth. Physical examination revealed symmetrical patches of hypopigmentation and depigmentation on the chest, abdomen, upper and lower extremities, with islands of normal pigmentation within them (Figures 1 and 2). A white forelock was observed (Figure 3). Bilateral cervical, axillary, and inguinal freckling and café-au-lait spots on the trunk were also noted (Figures 4 and 5). Learning problems were evident. The parents were non-consanguineous.
 |  |
| Figure 4 | Figure 5 |
|---|---|
| Figure 4. Axillary freckling Figure 5. Inguinal freckling | |
Routine laboratory tests were within normal values. Ophthalmologic, neurologic, orthopedic, and otologic evaluations were normal. Cerebral MRI and bone x-ray were normal.
The 33-year-old father presented with hypopigmented patches, axillary freckling and more than six café-au-lait spots greater than 15 mm. We were told the grandfather had similar patches. Genetic study of the father and child revealed a p.Gly610Asp mutation in the KIT gene, but no alteration for neurofibromatosis.
Discussion
Piebaldism is usually a benign isolated skin condition, but there are rare reports of piebaldism association with Hirschprung disease, mental retardation, NF1, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease, deafness, and cerebellar ataxia [1].
In this case the molecular study confirmed a mutation on the piebaldism gene, but clinical features also suggested the associated diagnosis of NF1, because of Crowe sign (axillary freckling) and the number and dimension of café-au-lait macules.
Inactivating mutations of the KIT gene results in decreased receptor tyrosine kinase signalling, impaired melanoblast development, and a decrease in melanogenesis. In contrast, inactivating mutations in the NF1 gene result in hyperactivation of the RAS proto-oncogene and enhanced tyrosine kinase receptor signalling [11].
At least four cases of piebaldism associated with neurofibromatosis have been reported and none of them developed neurofibromas, although at their young age (3-17 years of age) it is not possible to exclude the possibility of subsequent development of those lesions [2, 12, 13]. There is evidence that mast cells are implicated as an active participant in tumor formation and the activation of the c-kit receptor is required in the tumor microenvironment to allow neurofibroma progression. Considering that c-kit activity controls migration, proliferation, and survival of NF1+/- bone marrow-derived mast cells [11] and that schwann cells and fibroblasts, the two main components of neurofibromas, secrete KIT-ligand in response to different stimuli [11, 14], the low levels of mastocytes and c-kit in piebaldism may explain the lack of neurofibromas when NF1 is associated; the activation of the c-kit receptor seems to be crucial for the initiation and progression of neurofibromas [11].
Whether the simultaneous occurrence of these two dominantly inherited diseases is significantly higher than by chance remains to be established [14]. The pigment alterations in piebaldism usually remain stable, are permanent, and are unresponsive to medical treatments and phototherapy. However, in a small number of patients, repigmentation may occur spontaneously [1, 15]. Patients generally have a normal life span but some experience social disabilities requiring psychological support [1]. However, the diagnosis of piebaldism should raise the suspicion of neurofibromatosis, which requires adequate follow-up and genetic counseling.
To our knowledge the p.Gly610Asp of the exon 12 of the KIT gene mutation has never been reported in association with piebaldism [3, 4].
References
1. Thomas I, Kihiczak GG, Fox MD, Janninger CK, Schwartz RA. Piebaldism: an update. Int J Dermatol 2004;43:716-719. [PubMed]2. Angelo C, Cianchini G, Grosso MG, Zambruno G, Cavalieri R, Paradisi M. Association of piebaldism and neurofibromatosis type 1 in a girl. Pediatr Dermatol 2001;18:490-493. [PubMed]
3. Murakami T, Fukai K, Oiso N, Hosomi N, Kato A, Garganta C, Barnicoat A, Poppelaars F, Aquaron R, Paller AS, Ishii M. New KIT mutations in patients with piebaldism. J Dermatol Sci 2004;35:29-33. [PubMed]
4. Murakami T, Hosomi N, Oiso N, Giovannucci-Uzielli ML, Aquaron R, Mizoguchi M, Kato A, Ishii M, Bitner-Glindzicz M, Barnicoat A, Wilson L, Tsukamoto K, Ueda H, Mancini AJ, Suzuki T, Riley J, Miertus J, Camargo M, Santoro-Zea A, Atkin J, Fukai K. Analysis of KIT, SCF, and initial screening of SLUG in patients with piebaldism. J Invest Dermatol 2005;124:670-672. [PubMed]
5. Richards KA, Fukai K, Oiso N. A novel KIT mutation result in piebaldism with progressive depigmentation. J Am Acad Dermatol 2001;44:288-292. [PubMed]
6. Sanchez-Martin M, Perez-Losada J, Rodriguez-Garcia A, González-Sánchez B, Corp. BR, Kuster W, Moss C, Spritz RA, Sánchez-García I. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A 2003;122A:125-132. [PubMed]
7. Chow RK, Stewart WD, Ho VC. Graft-versus-host reaction affecting lesional skin but not normal skin in a patient with piebaldism. Br J Dermatol 1996;134:134-137. [PubMed]
8. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151:33-40. [PubMed]
9. National Institutes of Health Consensus Development Conference. Neurofibromatosis: conference statement. Arch Neurol 1988;45:575-578. [PubMed]
10. Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pveritz RE, Rubenstein A, Viskochil D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51-57. [PubMed]
11. Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW. Nf1-dependent tumors require a microenvironment containing Nf1+/- - and c-kit-dependent bone marrow. Cell 2008;135:437-448. [PubMed]
12. Chang T, McGrae JD, Hashimoto K. Ultrastructural study of two patients with both piebaldism and neurofibromatosis 1. Pediatr Dermatol 1993;10:224-234. [PubMed]
13. Tay YK. Neurofibromatosis 1 and piebaldism: a case report. Dermatology 1998;197:401-402. [PubMed]
14. Spritz RA, Itin PH, Gutmann DH. Piebaldism and neurofibromatosis type 1: horses of very different colors. J Invest Dermatol 2004;122:xxxiv-xxxv. [PubMed]
15. Matsunaga H, Tanioka M, Utani A, Miyachi Y. Familial case of piebbaldism with regression of white forelock. Clin Exp Dermatol 2008;33:511-512. [PubMed]
© 2010 Dermatology Online Journal