9p21 Deletions in Primary Melanoma
Published Web Location
https://doi.org/10.5070/D38nb5h28rMain Content
9p21 Deletions in Primary Melanoma
Mark F. Naylor, Sarah Brown, Christie Quinlan, Jan V. Pitha and Mark A. Everett
Dermatology Online Journal: 3(2): 1
AbstractFormalin-fixed, paraffin-embedded primary melanoma biopsies were evaluated for evidence of genomic loss on the short arm of chromosome 9 using microsatellite PCR assays for the D9S157, D9S161 and D9S171 loci. Paired normal and tumor DNA was extracted from the same block for comparison of microsatellite marker patterns. Some detectable abnormality was seen in at least one of these loci in 15 of 44 evaluable specimens (34%). Homozygous deletions were detectable at these loci in 8 of 44 informative specimens (18%) and hemizygous deletions were seen in 11 of 44 informative specimens (25%). Deletions at 9p were more likely to be found as primary tumor thickness increased (p < 0.05). This evidence supports the concept that 9p21 deletions are involved in primary sporadic melanomas, and that 9p deletions are not solely an in vitro phenomenon. |
IntroductionDeletions in the 9p21 region of chromosome 9 occur frequently in many human tumors, particularly gliomas [1, 2], lymphoid leukemias, lymphomas [2-4], head and neck tumors [5, 6] , esophageal carcinomas [7, 8], pancreatic adenocarcinomas [7, 9] bladder tumors [10, 11], lung cancers [2, 12-14], sarcomas, mesotheliomas [15, 16], and melanomas [2, 17-19]. A recently reported pan-genome survey looking for deletions in melanoma indicated that defects at the 9p21 locus were the most common chromosomal deletions seen in these tumors [18]. To better define the extent and frequency of 9p21 deletions in primary melanomas, formalin-fixed, paraffin-embedded human melanomas biopsies were examined for loss of heterozygosity at three 9p21 loci using PCR-based microsatellite assays.MethodsAn average of three, 40 µm sections of the paraffin-embedded blocks were cut on a microtome using disposable blades in a UV hood. The microtome was exposed to UVC 15-30 minutes prior to use. Sections were then carefully microdissected to separate tumor from normal tissues. Only areas that were clearly either normal tissue or tumor were used for DNA extractions. Microdissected areas were then deparaffinized and digested with proteinase K for 48-72 hours as described by Wright and Manos [20]. After proteinase K heat inactivation, the resulting aqueous phase was used as PCR target. Microsatellite assays used primers for the D9S157, D9S161 and D9S171 loci from Research Genetics, Inc. (Huntsville, AL). These markers were chosen empirically for their robustness in formalin-fixed DNA specimens. Polymerase chain reaction (PCR) conditions were established for optimum UV-visualization of primer products in ethidium bromide stained 2% agarose gels (NuSieve 3:1). Reactions were carried out on a model 480 thermal cycler (Perkin-Elmer, Norwalk, CT) using a 35 cycle protocol with 0.2 mM dNTPs, 50 mM KCl, 0.5 µCi of 33P-dATP, and 1.25 units of Taq DNA polymerase in a 20 µl volume (final concentrations). MgCl2 and primer concentrations for the three assays were 2 mM and 1.0 µM for the D9S157 assay, 2 mM and 1.5 µM for the D9S161 assay, and 2 mM and 0.5 µM for the D9S171 assay. Comparative PCR assays for paired tumor and normal samples were done simultaneously. PCR products from paired normal and tumor reactions were run in adjacent wells of a non-denaturing 15% polyacrylamide gel at 650 V and imaged with autoradiography. Visual assesment of autoradiograph band intensities were confirmed and quantitated in most cases with densitometry using a conventionally scanned image and image analysis software (NIH Image 1.58). Equivocal cases or those with saturated band intensities on autoradiography were evaluated directly from the gel using a phosphorimager (PhosphorImager SI, Molecular Dynamics, Inc.). Band intensities of normal alleles were used for baseline comparison values. A hemizygous allele loss was considered to be present in the tumor when a band was diminished in expected intensity by at least 50% on at least two separate PCR runs. |

|
Figure 1: Examples of a hemizygous D9S157 losses in specimens 59 and 60. In both of these cases, the upper allelic band (arrow) is diminished in the tumor samples (T59 and T60) relative to the expected intensity by >50ompared to the normal sample allele bands (N59 and N60). |
A homozygous allele loss was scored if the tumor alleles were repeatably absent or diminished from expected intensity by at least 90% while simultaneous control reactions from the same sample showed strong bands. Control reactions at uninvolved chromosome loci including D3S1038 and D18S44 (not frequently involved in reports of karyotypic and genomic abnormalities in melanoma), were run simultaneously with repeat 9p21 assays to rule out technical artifact. ResultsMicroscope slides of approximately 200-300 biopsies were screened for a combination of well separated tumor-bearing areas and adequate amounts of normal tissue. Approximately 75 of these biopsies were initially extracted and screened for target adequacy using control primers for exon 3 of carbonyl reductase (NADP3) designed to give a 194 bp product. 52 of 75 were found to give strong enough signal in both normal and tumor DNA extracts to warrant further evaluation. Usable data from at least one of the three loci was found in 44 of these 52 biopsies and these data was used to generate Figure 2.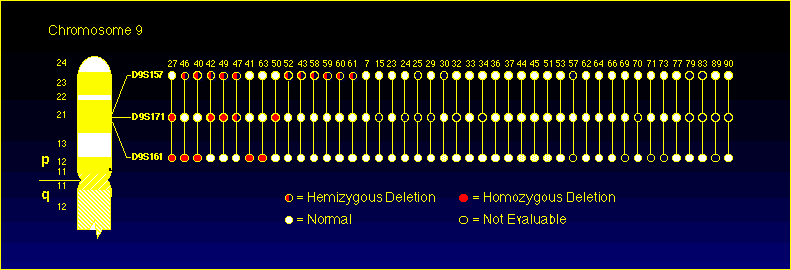
Figure 2: Allelic loss data for 44 primary melanomas. The short arm of chromosome 9 and the relative positions of the D9S157, D9S171 and D9S161 markers is depicted on the left side of the figure. Specimen numbers are indicated above the ideogram depiction of marker status. This summarizes results obtained in these specimens with the D9S157, D9S161 and D9S171 assays. Of the 44 evaluable specimens, 15/44 (34%) had detectable deletions (both hemizygous and homozygous), 8/44 (18%) had homozygous deletions and 11/44 (25%) had hemizygous deletions.
Table 1. Breslow's thickness data
Table 1 shows primary tumor thickness for the 36 biopsies for which this information was available. As can readily be seen from Table 1, the samples in this study were biased toward thicker lesions. This was necessary because of the physical requirement for adequate and well separated areas of normal and tumor cells in the same biopsy. When the average thickness of tumors with detectable 9p21 deletions at the three loci assayed were compared to those which did not have detectable deletions, the mean thickness was significantly greater in tumors with deletions than the mean thickness of tumors without 9p21 deletions (p < 0.05). ConclusionsThe 9p21 region has been identified as the site of at least two putative tumor suppressor genes, CDKN2A (p16), and CDKN2B (p15). These are kinase inhibitor genes that function to inhibit cell cycle transit [21-24]. Kamb et. al originally reported that the MTS1 open reading frame of CDKN2A was deleted or mutated in 750f 99 melanoma cell lines [23]. It has been reported that cultured tumor cells have a higher frequency of deletions at 9p21 than do primary tumors, probably because these deletions confer a growth advantage to cells in vitro [6, 25]. This has been interpreted by some to mean that CDKN2A deletion may principally be an in vitro phenomenon, implying a minor role in tumor biology in vivo and that CDKN2A is not involved in actual melanoma development.More recent evidence suggests that CDKN2A is indeed involved in melanoma development. Strong support for CDKN2A as a melanoma tumor suppressor comes from the fact that it is a familial tumor gene [21]. Additional indirect evidence is provided by the demonstration of CDKN2A mutations in melanoma cell lines consistent with those induced by ultraviolet light [26]. Perhaps the most direct evidence available so far to implicate this gene in melanoma development is the loss of CDKN2A expression in primary tumor material as shown by immunohistochemical staining [27] . This study confirms a relationship between tumor thickness and 9p21 deletions seen by others [17, 27], and suggests that 9p21 deletions most often develop during progression of the tumor. However, early lesions do show evidence of 9p21 abnormalities, implying that these can occur in the early evolution of the tumor. We found that 2/11 informative early lesions (e.g., Breslow thickness less than 1.5 mm) showed evidence of 9p21 deletion. Healy et. al., noted similar findings with 3/9 deletions at the 9p21 locus in primary tumors less than 1.5 mm in thickness [18] . This is not particularly surprising since at least one 9p21 gene (e.g. CDKN2A) is known to be a familial melanoma gene, implying that it can be the first genetic abnormality in a tumor [21, 28] . The present study reveals evidence of deletion in 430f evaluable cases of primary melanomas, similar to or slightly lower than other recent reports of loss of heterozygosity at this locus in melanomas [19, 29, 30] . These studies may however, actually underestimate abnormalities of 9p21 genes for several reasons. Since this is a study of primary lesions, and p16 inactivation may increase in frequency as melanomas progress from primary to metastatic tumors [27] . Formalin fixed specimens do not always yield interpretable data at each microsatellite locus assayed, so some of the loci in the specimens reported here as non-evaluable may actually be deletions. Small homozygous deletions, which are known to occur in this region [2,31, 32] would not necessarily be detected by this methodology. Chromosomal marker studies will not reveal examples of bilateral point mutations in causative tumor suppressor genes. We were unable to get enough reliable sequence data from these formalin fixed specimens to make any assessment of the frequency of CDKN2A mutations in these specimens. Finally, this approach is of no value in detecting abnormalities at the levels of protein interactions or protein expression. A good example of this is under expression due to methylation of promoter sequences, an abnormality known to occur with both CDKN2A and CDKN2B [33, 34] .The marker data presented here confirm that sporadic melanomas exhibit a substantial frequency of abnormalities in the p21 region of chromosome 9. The present study, in conjunction with other reports of frequent deletion of the 9p21 region in tumor material provides strong evidence that a gene or genes at this locus are causally linked to sporadic as well as familial melanomas. While this evidence does not prove that CDKN2A or CDKN2B are causative tumor suppressors in sporadic melanomas, these data is consistent with that concept. Acknowledgement:The authors acknowledge the Oklahoma Center for the Advancement of Science and Technology for supporting this work, the Molecular Biology Resource Core Laboratory in Oklahoma City and the Oklahoma Center for Molecular Medicine (OCMM) Computer Facility in Norman, whose resources were used in the completion of this work. References1. Moulton T, Samara G, Chung WY, et al.: MTS1/p16/CDKN2 lesions in primary glioblastoma multiforme. Am J Pathol 146(3):613-619, 1995.2. Coleman A, Fountain JW, Nobori T, et al.: Distinct deletions of chromosome 9p associated with melanoma versus glioma, lung cancer, leukemia. Cancer Res 54(2):344-348, 1994. 3. Ogawa S, Hirano N, Sato N, et al.: Homozygous loss of the cyclin-dependent kinase 4-inhibitor (p16) gene in human leukemias. Blood 84(8):2431-2435, 1994. 4. Duro D, Flexor MA, Bernard O, et al.: Alterations of the putative tumor suppressor gene p16/MTS1 in human hematological malignancies. Comptes Rendus de l Academie des Sciences - Serie Iii, Sciences de la Vie 317(10):913-919, 1994. 5. Lydiatt WM, Murty VV, Davidson BJ, et al.: Homozygous deletions and loss of expression of the CDKN2 gene occur frequently in head and neck squamous cell carcinoma cell lines but infrequently in primary tumors. Genes, Chromosomes & Cancer 13(2):94-98, 1995. 6. Zhang S-Y, Klein-Szanto AJP, Sauter ER, et al.: Higher frequency of alteration in the p16/CDKN2 gene in squamous cell carcinoma cell lines than in primary tumors of the head and neck. Cancer Res 54(19):5050-5053, 1994. 7. Liu Q, Yan YX, McClure M, et al.: MTS-1 (CDKN2) tumor suppressor gene deletions are a frequent event in esophagus squamous cancer and pancreatic adenocarcinoma cell lines. Oncogene 10(3):619-622, 1995. 8. Mori T, Miura K, Aoki T, et al.: Frequent somatic mutation of the MTS1/CDK4I (multiple tumor suppressor/cyclin-dependent kinase 4 inhibitor) gene in esophageal squamous cell carcinoma. Cancer Res 54(13):3396-3398, 1994. 9. Caldas C, Hahn SA, Costa LTd, et al.: Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 8(1):27-32, 1994. 10. Gonzalez-Zulueta M, Shibata A, Ohneseit P, et al.: High frequency of chromosome 9p allelic loss and CDKN2 tumor suppressor gene alterations in squamous cell carcinoma of the bladder. J Natl Cancer Inst 87(18):1383-1393, 1995. 11. Stadler WM, Sherman J, Bohlander SK, et al.: Homozygous deletions within chromosomal bands 9p21-22 in bladder cancer. Cancer Res 54(8):2060-2063, 1994. 12. Xiao S, Li D, Corson JM, et al.: Codeletion of p15 and p16 Genes in Primary Non-Small Cell Lung Carcinoma. Cancer Res 55(14):2968-2971, 1995. 13. Mead LJ, Gillespie MT, Irving LB, et al.: Homozygous and hemizygous deletions of 9p centromeric to the interferon genes in lung cancer. Cancer Res 54(9):2307-2309, 1994. 14. Merlo A, Gabrielson E, Askin F, et al.: Frequent loss of chromosome 9 in human primary non-small cell lung cancer. Cancer Res 54(3):640-642, 1994. 15. Maelandsmo GM, Berner JM, Florenes VA, et al.: Homozygous deletion frequency and expression levels of the CDKN2 gene in human sarcomas--relationship to amplification and mRNA levels of CDK4 and CCND1. Br J Cancer 72(2):393-398, 1995. 16. Cheng JQ, Jhanwar SC, Lu YY, et al.: Homozygous deletions within 9p21-p22 identify a small critical region of chromosomal loss in human malignant mesotheliomas. Cancer Res 53(20):4761-4764, 1993. 17. Healy E, Rehman I, Angus B, et al.: Loss of heterozygosity in sporadic primary cutaneous melanoma. Genes, Chromosomes & Cancer 12(2):152-156, 1995. 18. Healy E, Belgaid CE, Takata M, et al.: Allelotypes of primary cutaneous melanoma and benign melanocytic nevi. Cancer Res 56(3):589-593, 1996. 19. Walker GJ, Palmer JM, Walters MK, et al.: Refined localization of the melanoma (MLM) gene on chromosome 9p by analysis of allelic deletions. Oncogene 9(3):819-824, 1994. 20. Wright DK, Manos MM, Sample preparation from paraffin-embedded tissues, in PCR Protocols, M.A. Innis, et al., Editor. 1990, Academic Press, Inc.: San Diego. p. 153-158. 21. Naylor MF, Quinlan C, Everett MA: Involvement of the p16INK4 (CDKN2) gene in familial melanoma. Melanoma Res in press, 1996. 22. Nobori T, Miura K, Wu DJ, et al.: Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368(6473):753-756, 1994. 23. Kamb A, Gruis NA, Weaver-Feldhaus J, et al.: A cell cycle regulator potentially involved in genesis of many tumor types. Science 264(5157):436-440, 1994. 24. Serrano M, Hannon GJ, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366(6456):704-707, 1993. 25. Spruck CH, Gonzalez-zuleta M, Shibata A, et al.: p16 gene in uncultured tumours. Nature 370(6486):183-184, 1994. 26. Pollock PM, Yu F, Qiu L, et al.: Evidence for u.v. induction of CDKN2 mutations in melanoma cell lines. Oncogene 11(4):663-668, 1995. 27. Reed JA, Loganzo Jr. F, Shea CR, et al.: Loss of Expression of the p16/Cyclin-dependent Kinase Inhibitor 2 Tumor Suppressor Gene in Melanocytic Lesions Correlates with Invasive Stage of Tumor Progression. Cancer Res 55(13):2713-2718, 1995. 28. Walker GJ, Hussussian CJ, Flores JF, et al.: Mutations of the CDKN2/p16INK4 gene in Australian melanoma kindreds. Hum Mol genet 4(10):1845-1852, 1995. 29. Puig S, Ruiz A, Lazaro C, et al.: Chromosome 9p deletions in cutaneous malignant melanoma tumors: the minimal deleted region involves markers outside the p16 (CDKN2) gene. Am J Hum Genet 57(2):395-402, 1995. 30. Holland EA, Beaton SC, Edwards BG, et al.: Loss of heterozygosity and homozygous deletions on 9p21-22 in melanoma. Oncogene 9(5):1361-1365, 1994. 31. Glendening JM, Flores JF, Walker GJ, et al.: Homozygous loss of the p15INK4B gene (and not the p16INK4 gene) during tumor progression in a sporadic melanoma patient. Cancer Res 55(23):5531-5539, 1995. 32. Fountain JW, Karayiorgou M, Ernstoff MS, et al.: Homozygous deletions with human chromosome band 9p21 in melanoma. Proc Natl Acad Sci 89(21):10557-10561, 1992. 33. Herman JG, Jen J, Merlo A, et al.: Hypermethylation-associated inactivation indicates a tumor suppressor role for the p15INK4B1. Cancer Res 56(4):722-727, 1996. 34. Herman JG, Merlo A, Mao L, et al.: Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 55(20):4525-4530, 1995. 35. Weber J, May P: Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 44(3):388-396, 1989. 36. Breslow A: Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Annals of Surgery 172(5):902-8, 1970. 37. Breslow A: Tumor thickness, level of invasion and node dissection in stage I cutaneous melanoma. Annals of Surgery 182(5):572-5, 1975. 38. Wanebo HJ, Fortner JG, Woodruff J, et al.: Selection of the optimum surgical treatment of stage I melanoma by depth of microinvasion: Use of the combined microstage technique (Clark-Breslow). Annals of Surgery 182(3):302-15, 1975. 39. Hansen MG, McCarten AB: Tumor thickness and lymphocytic infiltration in malignant melanoma of the head and neck. American Journal of Surgery 128(4):557-61, 1974. 40. Sober AJ, Mihm MC, fitzpatrick TB, et al., Malignant melanoma of the skin,and benign neoplasms and hyperplasias of melanocytes in the skin, in Dermatology in General Medicine, T.B. Fitzpatrick, et al., Editor. 1987, McGraw-Hill: New York. p. 947-966. 41. Eldh J, Boeryd B, Peterson LE: Prognostic factors in cutaneous malignant melanoma in stage I. A clinical, morphological and multivariate analysis. Scandinavian Journal of Plastic & Reconstructive Surgery 12(3):243-55, 1978. 42. Balch CM, Soong SJ, Murad TM, et al.: A multifactorial analysis of melanoma. II. Prognostic factors in patients with stage I (localized) melanoma. Surgery 86(2):343-51, 1979. 43. Sober AJ, Mihm MC, fitzpatrick TB, et al., Malignant melanoma of the skin,and benign neoplasms and hyperplasias of melanocytes in the skin, in Dermatology in General Medicine, T.B. Fitzpatrick, et al., Editor. 1979, McGraw-Hill: New York. p. 629-654. 44. Morgan DO: Principles of CDK regulation. Nature 374:131-134, 1995. 45. Hunter T, Pines J: Cyclins and cancer II: cyclin D and CDK inhibitors come of age. Cell 79(4):573-582, 1994. 46. Guan KL, Jenkens CW, Li Y, et al.: Growth suppression by p18, a p16INK4MTS1- and p14INK4MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes and Development 8:2939-2952, 1994. 47. Peters G: Stifled by inhibitions. Nature 371(6494):204-205, 1994. 48. Halevy O, Novitch BG, Spicer DB, et al.: Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 267(5200):1018-1021, 1995. 49. Parker SB, Eichele G, Zhang P, et al.: p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 267(5200):1024-1027, 1995. 50. Hannon GJ, Beach D: p15INK4B is a potential effector of TGF-b-induced cell cycle arrest. Nature 371(6494):257-261, 1994. 51. Yeager T, Stadler W, Belair C, et al.: Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer Res 55(3):493-497, 1995. 52. Hartwell LH, Kastan MB: Cell cycle control and cancer. Science 266(5192):1821-1828, 1994. 53. Herman JG, Latif F, Weng Y, et al.: Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci 91(21):9700-9704, 1994. 54. Deobagkar DD, Liebler M, Graessmann M, et al.: Hemimethylation of DNA prevents chromatin expression. Proc Natl Acad Sci 87:1691-1695, 1990. 55. Gonzalez-Zulueta M, Bender CM, Yang AS, et al.: Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 55(20):4531-4535, 1995. |
© 1997 Dermatology Online Journal for use in electronic version, otherwise copyright retained by the author(s) |