Lipoid proteinosis: Report of three siblings
Published Web Location
https://doi.org/10.5070/D38m1545svMain Content
Lipoid proteinosis: Report of three siblings
Soheila Nasiri MD, Nima Sarrafi-rad MD, Sima Kavand MD, Marjan Saeedi MD
Dermatology Online Journal 14 (11): 6
Skin Research Center, Shahid Beheshti University, M.C. Shohada-e Tajrish Hospital, Tehran, Iran. marjan406@yahoo.comAbstract
Lipoid proteinosis is a very rare genodermatosis characterized by infiltration of hyaline material into the skin, oral cavity, larynx and internal organs. It usually presents in infancy with hoarseness. Although about 300 cases have been reported in the literature, the occurrence of the disease is rare in siblings. In this report we introduce three siblings with this disease.
Introduction
Lipoid proteinosis (LP) is a rare disorder that presents in early childhood with hoarseness, skin infiltration and thickening, beaded papules on eyelid margins, and facial acneiform or pock-like scars. Other organs such as the respiratory system are sometimes involved. Histological examinations show an extracellular PAS-positive hyaline material. The disorder is attributed to a mutation of the extracellular-matrix protein-1 located on chromosome 1q21 [1].
Clinical synopsis
Three siblings, two girls (3- and 6-years-old) and a 9-year-old boy born of consanguineous parents, came to our clinic with hoarse voices and multiple, asymptomatic, raised skin lesions present since early childhood. All three had a normal appearance and cry at birth, but started developing hoarseness of the voice (as noticed by their mother) by the end of the first year. The 6-year-old girl had more severe involvement. She had hoarseness that was noted as a faint or weak cry and oral erosion since early infancy. Her lesions appeared in two stages. The first stage manifested with vesicles and erosions mimicking epidermolysis bullosa. The vesicles healed with hemorrhagic crusting and resultant linear scarring. Skin fragility and easy wounding with minor trauma was noted. In the second stage beaded papules along her eyelid margins appeared. Generalized skin thickening with a waxy, yellow appearance was noted and thickening of the frenulum of tongue resulted in restriction in the ability to protrude the tongue. Hyperkeratosis developed over areas subjected to repeated minor trauma. Pock-like or acneiform scars on the face and extremities were noted on physical exam (Figs. 1 & 2). The other two siblings had similar, but milder, symptoms (Figs. 3 - 6). Other cutaneous surfaces and mucosal membranes were normal on clinical examination. Systemic examinations including CNS surveys were normal. Routine laboratory data were reported to be normal in all three patients.
 |  |
| Figure 1 | Figure 2 |
|---|---|
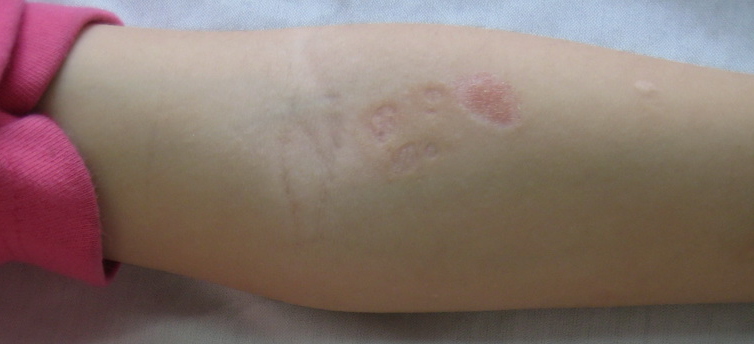
| Figure 1. Pock-like scars on extremities of the 6-year-old girl Figure 2. Erosions and hemorrhagic crusts on the back of the 6-year-old girl | |
 |  |
| Figure 3 | Figure 4 |
|---|---|
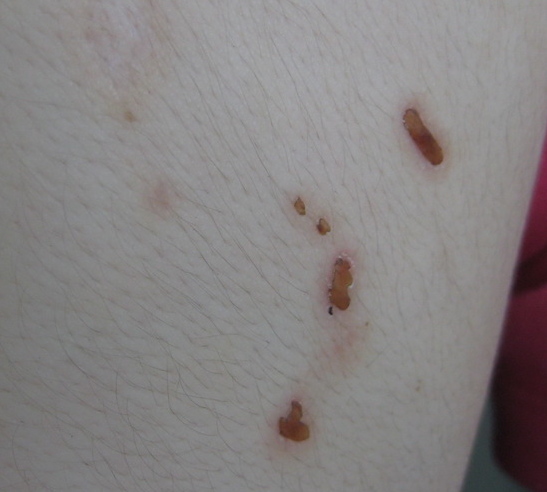
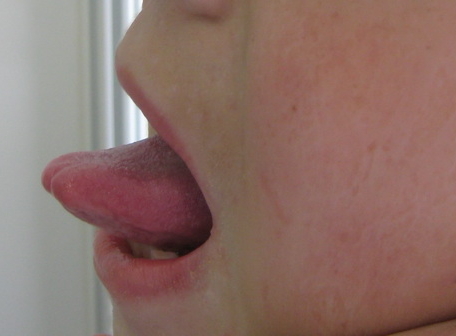
| Figure 3. Restricted mobility of the tongue due to infiltration of the frenulum in 9-year-old boy Figure 4. Pock-like scars and crusts on the face of the 9-year-old boy | |
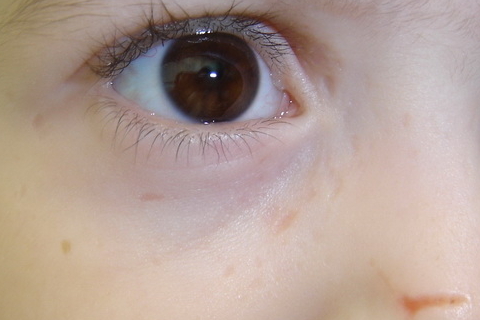 | 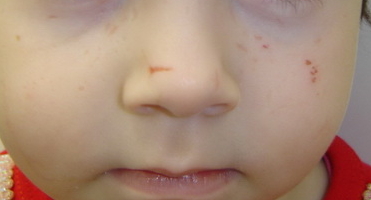 |
| Figure 5 | Figure 6 |
|---|---|
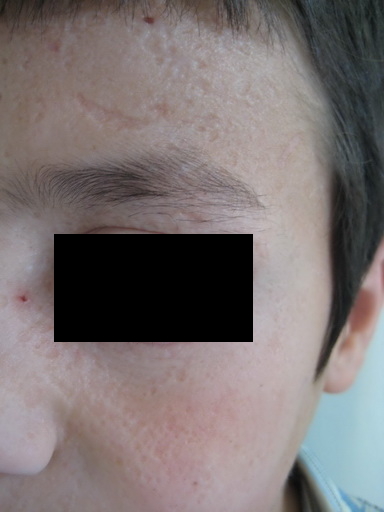
| Figure 5. Pock-like scars on face and Beaded papule on the eyelid of the 3-year-old girl Figure 6. Crusted erosions leading to linear scars on the face of the 3-year-old girl | |
Microscopic findings
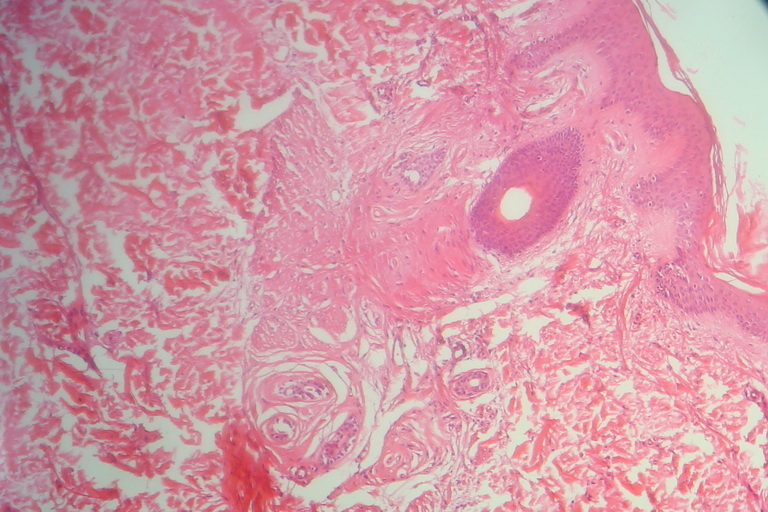
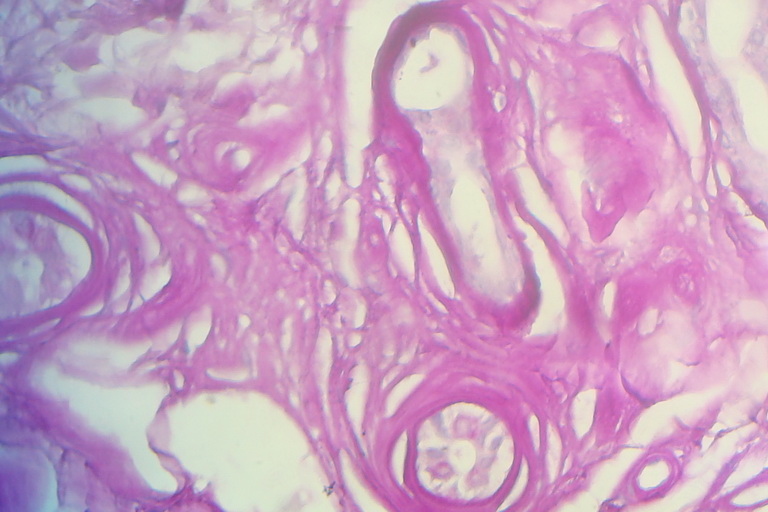
Histological examinations of representative skin lesions from both siblings showed deposition of a pink, amorphous material in the papillary dermis, around blood vessels, and in appendages (Fig. 7). These deposits stained positive with PAS stain (Fig. 8).
 |  |
| Figure 7 | Figure 8 |
|---|---|
| Fig 7. Deposition of eosinophilic hyaline material in the papillary dermis and deeper part of dermis (H&E x10) Fig 8. Deposition of PAS positive hyaline material in the dermis of a scar-like lesion (PAS x40) | |
Discussion
Urbach and Wiethe first described the lipoid proteinosis in Germany in 1929, but it is estimated that 25 percent of cases occur in South Africa, particularly in the Namaqualand region in the towns of Concordia, Okiep, Nababeep, and Springbok. This is a result of the "founder effect," whereby a condition, rare in the original population, is carried to a new area by migration of one or more individuals within a small group. The abnormality therefore becomes more common in the new population. The original carrier of this condition has been found to be one Jacob Cloete, a German immigrant to the Cape in 1652. His great-grandson Gerrit Cloete migrated to Namaqualand in 1742. Because the area is somewhat isolated, consanguineous marriages were relatively common, leading to a relatively high proportion of homozygous affected individuals [2].
Lipoid proteinosis is inherited in an autosomal recessive fashion and is caused by mutations in the gene encoding extracellular matrix protein-1 (ECM-1). The majority of mutations occur in exon 7. ECM-1 that is known to inhibit bone mineralization, contributes to epidermal differentiation and stimulates angiogenesis [1].
The most reliable clinical signs for diagnosing lipoid proteinosis are a hoarse voice and thickened sublingual frenulum, which prevents a patient from protruding the tongue. Secondary features include beaded eyelid papules, infiltration, warty papules of the skin around the elbow and extensor forearms, and mild alopecia. Increased scaring and photoaging of sun-exposed skin may be seen. Scars or scar-like lesions are present in areas of minor trauma. Central nervous system involvement is a well recognized, but variable feature of lipoid proteinosis. Amygdala dysfunction has been described, which leads to abnormal perception and appraisal of fear. Other neurologic and psychiatric findings include seizures, memory deficit, social and behavioral changes, paranoid symptoms, mental retardation, and aggressiveness. The abnormal deposition of hyaline material has been detected histopathologically in many internal organs, although this is typically asymptomatic [1]. Rare manifestations include: a localized form on both hands and wrists [3], an unusual presentation with verruca vulgaris [4], and forms that exhibit bilateral lens subluxation [5] and pseudomembranous conjunctivitis [6].
Histologically, PAS positive, diastase resistant thickening is seen at the dermal-epidermal junction, perivascular areas, and along adnexal epithelia; hyaline material may be noted in the dermis [1].
Lipoid proteinosis needs to be differentiated from erythropoietic protoporphyria (EPP), a condition characterized by skin involvement confined to sun-exposed areas and associated with photosensitivity. Erythropoietic protoporphyria also has a deposition of PAS-positive material; however it is less dense around blood vessels and never occurs around sweat glands [7].
Although 250 cases have been reported to date, occurrence of the disease in siblings is very rare. The most prevalent familial cases were reported in a family with four siblings (two brothers and two sisters) from a non-consanguineous marriage. All cases showed hoarseness and three of them presented the typical skin lesions with the classic appearance on optical and electronic microscopic evaluation [8]. One report of lipoid proteinosis in a brother and sister found only eyelid papules with no other sign, but the histological evaluation was classic for this disease [9]. Three familial cases of lipoid proteinosis involving a brother and sister and their nephew also had been reported from Iran [10]. In another report from India one brother and sister with the classical manifestation of this disease were introduced [7].
In conclusion, this report features one of the rare forms of lipoid proteinosis involving three siblings.
References
1. Dyer J.A. lipoid Proteinosis; Skin in nutritional, metabolic, and heritable Disease. In Fitzpatrick's Dermatology in general medicine (Wolf K, Goldsmith L, Katz S, et al), 7th edn, New York: Mc Graw Hill, 2008:1288-12922. Gordon H, Gordon W, Botha V. Lipoid proteinosis in an inbred Namaqualand community. Lancet. 1969 May 24; 1(7604):1032-5. PubMed
3. Hu S, Kuo TT, Hong HS. Lipoid proteinosis: report of a possible localized form on both hands and wrists. Int J Dermatol. 2005 May; 44(5):408-10. PubMed
4. Parlak AH, Koybasi S, Boran C, Ibrahimbas Y. Lipoid proteinosis: an unusual presentation with verruca vulgaris. J Dermatol. 2005 Sep; 32(9):751-5. PubMed
5. Mandal S, Dutta P, Venkatesh P, Sinha R, Kukreja M, Garg S. Bilateral lens subluxation in a case of lipoid proteinosis. J Cataract Refract Surg. 2007 Aug; 33(8):1469-70. PubMed
6. Barthelemy H, Mauduit G, Kanitakis J, Cambazard F, Thivolet J. Lipoid proteinosis with pseudomembranous conjunctivitis. J Am Acad Dermatol. 1986 Feb; 14(2 Pt 2):367-71. PubMed
7. Sethuraman G, Tejasvi T, Khaitan BK, Handa KK, Rao S, Singh MK, Sirka C, Sharma VK.Lipoid proteinosis in two siblings: a report from India. J Dermatol. 2003 Jul; 30(7):562-5. PubMed
8. Nanda A, Alsaleh QA, Al-Sabah H, Ali AM, Anim JT. Lipoid proteinosis: report of four siblings and brief review of the literature. Pediatr Dermatol. 2001 Jan-Feb; 18(1):21-6. PubMed
9. Sharma V, Kashyap S, Betharia SM, Gupta S, Pathak H. Lipoid proteinosis: a rare disorder with pathognomonic lid lesions. Clin Experiment Ophthalmol. 2004 Feb; 32(1):110-2. PubMed
10. Ehsani AH, Ghiasi M, Robati RM. Lipoid proteinosis: report of three familial cases. Dermatol Online J. 2006 Jan 27; 12(1):16. PubMed
© 2008 Dermatology Online Journal