X-Linked ocular albinism; Nettleship-Falls ocular albinism
Published Web Location
https://doi.org/10.5070/D38dz8n8rkMain Content
X-Linked ocular albinism; Nettleship-Falls ocular albinism
Alexandria V Booth MD, Anthony C Soldano MD, Jonathan Levine MD, Miriam Pomeranz MD
Dermatology Online Journal 14 (5): 4
Department of Dermatology, New York UniversityAbstract
A 39-year-old man with foveal hypoplasia, nystagmus, and decreased visual acuity was found to have multiple, cutaneous, hypopigmented macules. Macromelanosomes were demonstrated in normal skin on histopathologic examination. The patient's constellation of findings along with a strong X-linked inheritance pattern in family members led to the diagnosis of X-linked ocular albinism, which is an uncommon condition that is characterized by congenital nystagmus, iris translucency, hypopigmentation of the ocular fundus, strabismus, foveal hypoplasia, photophobia, and impaired vision.
Clinical synopsis
A 39-year-old man was referred to the Bellevue Hospital Center dermatology clinic by his ophthalmologist for a cutaneous examination. The patient originally presented for congenital nystagmus and lifelong reduced visual acuity. A maternal uncle and 2 of 4 brothers also have nystagmus with decreased visual acuity. Two sisters have unaffected vision; one sister has a son with vision problems similar to those of the patient. All family members live in the patient's native country of Mali.
Physical examination
Numerous, scattered, hypopigmented macules were distributed over the chest, back, buttocks, upper extremities, and lower extremities. Generalized scale was noted with polygonal scale over the lower extremities. Nystagmus with foveal hypoplasia was present; visual acuity was 20/100.
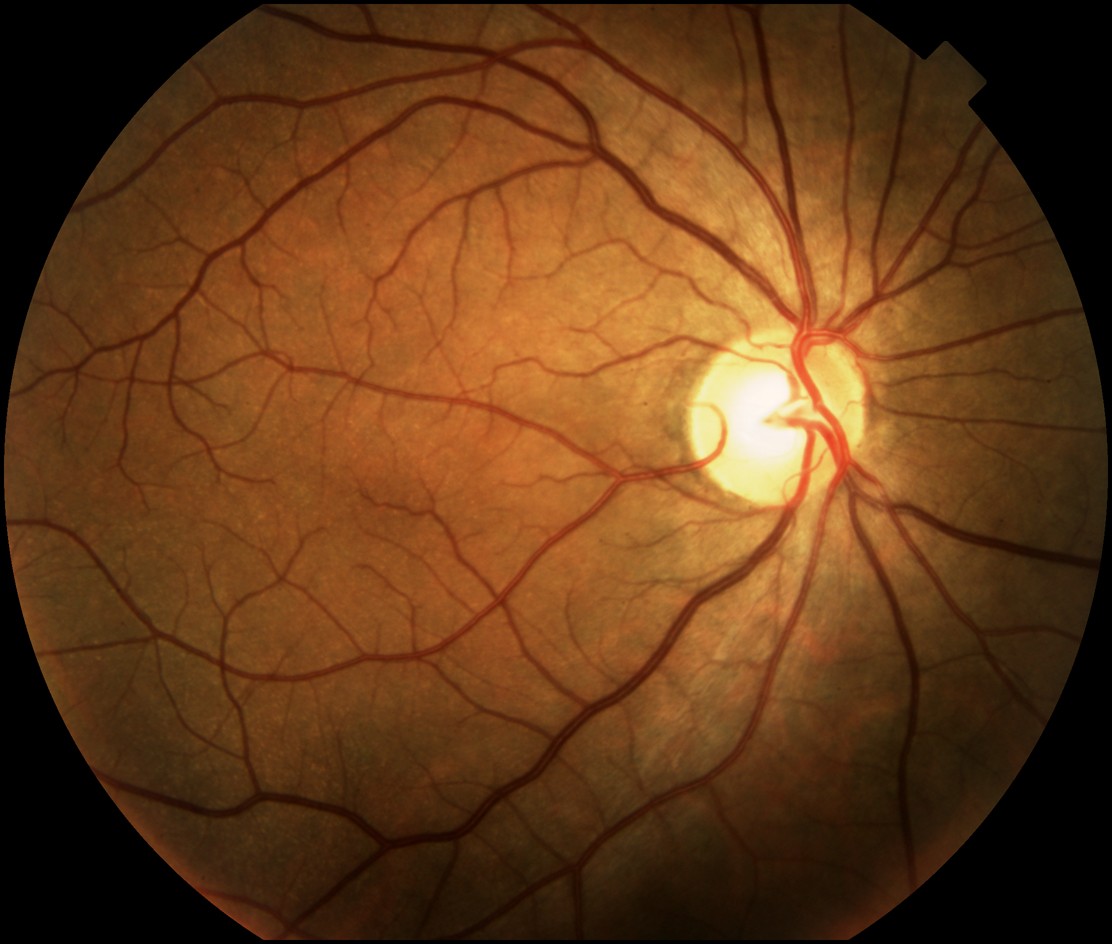 | 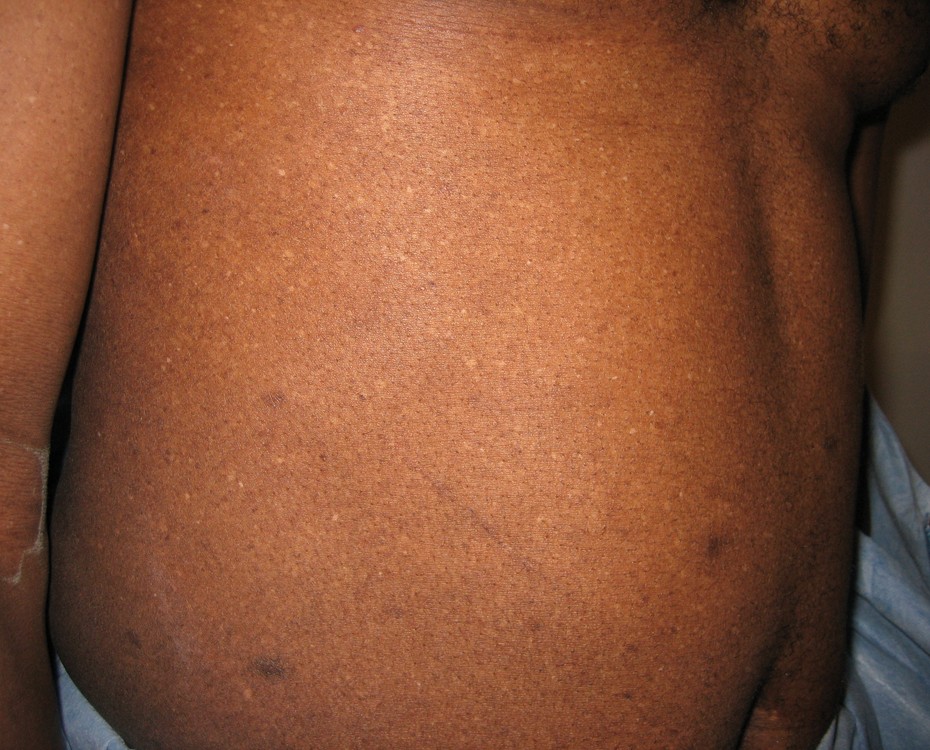 |
| Figure 1 | Figure 2 |
|---|
Laboratory data
None
Histopathology
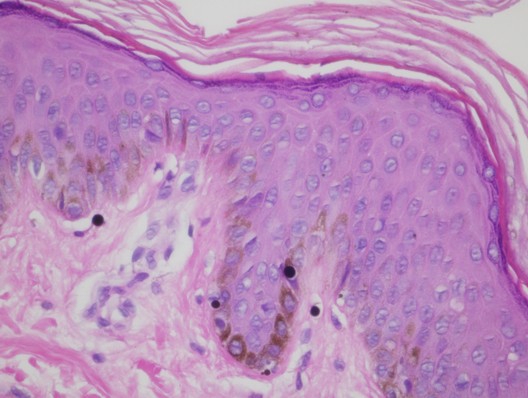 |
| Figure 3 |
|---|
There is slight hyperpigmentation of the epidermal basal layer with an average normal number of melanocytes. Large melanosomes are present within the cytoplasm of both melanocytes and basal keratinocytes.
Comment
Ocular albinism type 1 (OA1) occurs primarily in XY males and is characterized by congenital nystagmus, iris translucency, hypopigmentation of the ocular fundus, strabismus, foveal hypoplasia, photophobia, and impaired vision [1]. Female carriers usually have normal vision with partial iris translucency and a classic mosaic pattern of retinal pigmentation [2, 3]. Skin and hair color are classically slightly lighter than are those of unaffected family members, compensating for age and ethnicity [3]. Affected individuals and some carriers may have cutaneous hypopigmented macules and patches that do not tan normally, which are more prominently observed in darkly-pigmented individuals [2, 3]. Light microscopy of normal skin or retinal pigment demonstrates macromelanosomes [2]. The presence of skin macromelanosomes is specific for OA1, is not obligate in men with OA1, and only occurs in a fraction of female carriers [1]. The OA1 gene was cloned in 1995 and has been mapped to the Xp 22.3-p22.2 region [3, 4]. About 48 percent of the reported mutations in the OA1 gene are iatrogenic deletions, and about 43 percent are point mutations [3]. It is almost exclusively expressed in pigmented tissue, such as retina, melanocytes, and melanoma cells [5]. The gene product has been characterized as a membrane glycoprotein that is localized to melanosomes, but its exact function in melanogenesis is unknown [1]. It has been reported that OA1 may play a role in regulating the late endosome/lysosomal compartment; the absence of OA1 results in continued vesicular traffic to mature melanosomes, with the subsequent development of a large vesicle that is filled with macromelanosomes at the expense of normal melanosomes [5]. There is close genetic linkage between OA1 and the X-linked ichthyosis steroid sulfatase gene on Xp22.32, with a few reported cases due to a contiguous gene defect [6, 7, 8]. Diagnostic testing for X-linked ocular albinism is not currently available in the United States. One proposed testing protocol, which employs multiplex polymerase chain reaction analysis, mutation detection with denaturing high-performance liquid chromatography, and sequence analysis, reports nearly 100 percent specificity and sensitivity [3].
References
1. Rosenberg T, et al. X-linked ocular albinism: prevalence and mutations- a national study. Eur J Hum Genet 1998;6:5702. O'Donnell FE, et.al. X-linked ocular albinism in Blacks. Ophthalmology 1978;96:1189
3. Heidge M, et al. Diagnostic testing for X-linked ocular albinism (OA1) with a hierarchical mutation screening protocol. Genet Test 2002;6:7
4. Bassi MT, et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nature Genet 1995;10:13
5. Shen B, et al. Intracellular and late endosomal effects of the ocular albinism type 1 gene product: consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2001;2:202
6. Hill VA, et al. Non-bullous congenital icthyosiform erythroderma, with ocular a albinism and Noonan syndrome. Clin Exp Dermatol 2000;25:611
7. Schnur RE, et al. An XP22 microdeletion associated with ocular albinism and icthyosis: approximation of breakpoints and estimation of deletion size by using cloned DNA probes and flow cytometry. Am J Hum Genet 1989;45:706
8. Sunohara N, et al. A new syndrome of anosomnia, icthyosis, hypogonadism, and various neurological manifestations with deficiency of steroid sulfatase and arylsulfatase C. Ann Neurol 1986;19:174
© 2008 Dermatology Online Journal