Juvenile amyopathic dermatomyositis
Published Web Location
https://doi.org/10.5070/D37wm4z0fxMain Content
Juvenile amyopathic dermatomyositis
J Scott Henning DO, Agnieszka Witkiewicz MD, Julie V Schaffer MD, and Seth J Orlow MD PhD
Dermatology Online Journal 11 (4): 11
Department of Dermatology, New York University School of Medicine
Abstract
A 3-year-old girl presented with a 6-month history of multiple, light-pink, flat-topped papules over the dorsal aspects of the metacarpophalangeal and interphalangeal joints of the hands and feet. Nailfold telangiectases, ragged cuticles, and a heliotrope color of the upper eyelids were also evident, but there was no clinical evidence of muscle weakness and levels of muscle enzymes were normal. A biopsy specimen from one of the papules showed a vacuolar interface dermatitis consistent with a diagnosis of dermatomyositis. This report draws attention to juvenile amyopathic dermatomyositis, which is an uncommon subtype of dermatomyositis with an excellent prognosis.
A 3-year-old girl presented to the Dermatologic Associates at New York University Medical Center in February 2005 for the evaluation of asymptomatic, flat-topped papules on the dorsal aspects of the hands and feet. The lesions previously had been diagnosed as flat warts, but there had been no response to a variety of topical therapies. The patient was healthy and active, with no recent fevers, malaise, weakness, myalgias, arthralgias, difficulty swallowing, cough, dyspnea, or photosensitivity.
Multiple, 2-3 mm, light-pink, flat-topped papules were present symmetrically over the dorsal aspects of the metacarpophalangeal and interphalangeal joints of the hands and feet. Similar papules were also noted over the sides and flexor aspects of the interphalangeal joints of the fingers. Dilated nailfold capillaries and ragged cuticles were evident, and the upper eyelids exhibited violaceous erythema. There were no lesions on the elbows, knees, scalp, or oral mucosa. Proximal muscle strength was 5/5 throughout, and there was no tenderness or swelling of the joints.
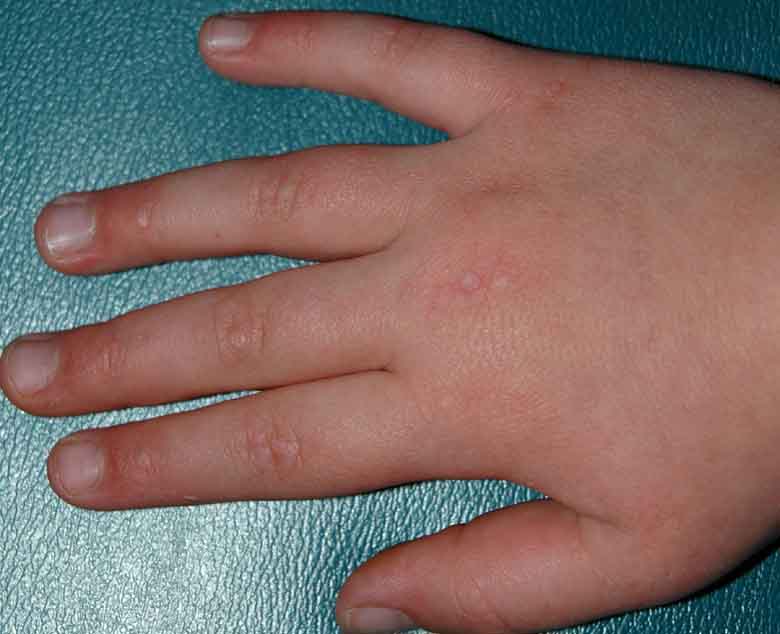
|
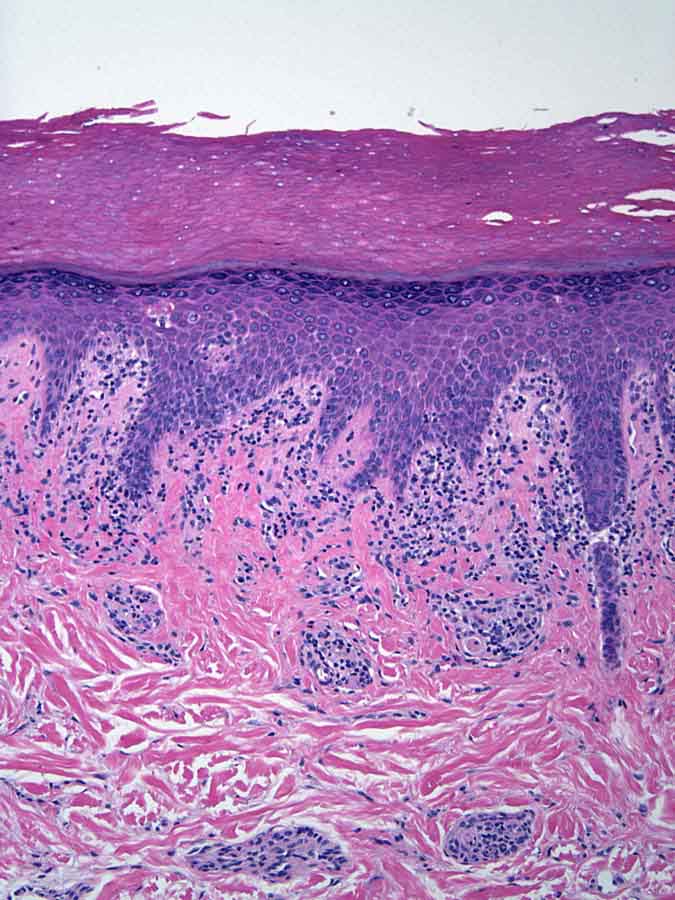
|
| Figure 1 | Figure 2 |
|---|
Serum creatine kinase, aldolase, lactate dehydrogenase, aspartate aminotranferase, and alanine aminotransferase levels, a complete blood count, and erythrocyte sedimentation rate were normal. Antinuclear antibodies were present at a titer of 1:160 with a speckled pattern.
Histopathology reveals a superficial and deep, perivascular, predominantly lymphocytic infiltrate. Lymphocytes extend to the dermoepidermal junction where there is vacuolar alteration of the basal layer and rare necrotic keratinocytes. A colloidal iron stain shows deposits of connective-tissue mucin.
Comment
Amyopathic dermatomyositis (ADM), a term coined by Pearson in 1979, is a subtype of dermatomyositis (DM), in which biopsy-confirmed, hallmark cutaneous manifestations of DM are present for at least 6 months without the development of either muscle weakness or elevated serum muscle enzyme levels [1, 2]. ADM accounts for approximately 5-10 percent of all cases of DM [3, 4, 5]. Euwer and Sontheimer [6] suggest that a diagnosis of ADM be further qualified as provisional (skin disease lasting for 6-24 months) or confirmed (skin disease lasting greater than 24 months). If more extensive muscle evaluation (e.g., magnetic resonance imaging, electromyogram, or muscle biopsy) is performed, the results must be normal in order to make a diagnosis of ADM. When such studies show evidence of subclinical myositis, patients are classified as having hypomyopathic dermatomyositis [2].
Juvenile DM has a bimodal age distribution, with peaks in incidence at ages 4-5 and 12-13 years [7]. Like the adult variant, the female-to-male ratio in juvenile DM is approximately 2:1 [7, 8]. Importantly, in contrast to adult DM, juvenile DM is not associated with an increased risk for the development of internal malignant conditions.
Classic heliotrope lesions and Gottron's papules are evident in one-half of patients with juvenile DM, while Gottron's sign (macular violaceous erythema on the extensor surfaces, with or-without associated scale) and periungual erythema are observed in more than three-quarters of cases [8]. Pruritus, scalp dermatitis, and facial edema (especially of the periorbital area) represent additional common cutaneous manifestations [8]. Calcinosis of the skin and muscle occurs in as many as 30-70 percent of children with DM, particularly in advanced disease or when the institution of systemic therapy is delayed, compared to fewer than 5 percent of adults with DM [5]. Other mucocutaneous findings occasionally associated with juvenile DM include hypertrichosis, gingival telangiectases, and panniculitis leading to lipoatrophy [5, 9, 10]. Systemic manifestations, such as nonerosive polyarthritis and interstitial lung disease, can occur in both classic and amyopathic forms of juvenile DM [8, 11].
Epidemiologic studies have documented an upper respiratory or gastrointestinal illness in the previous 3 months in a higher proportion of patients with new-onset juvenile DM than in age-matched controls, which supports a role for molecular mimicry in the pathogenesis of the disorder [7]. The resultant process of antigen-driven autoimmunity includes a dynamic interaction between muscle, blood vessels, and the immune system in a genetically susceptible individual (e.g., DQA1*0501-positive) [12]. A polymorphism in the tumor necrosis factor-α (TNFα) promoter that leads to increased production of both TNFα and thrombospondin-1 (an anti-angiogenic mediator that may contribute to vascular occlusion in DM) has been associated with a prolonged disease course in juvenile DM [13].
In children with DM, invasive diagnostic procedures such as electromyography and muscle biopsy tend to be avoided [14]. It is important to recognize that serum creatine kinase and aldolase levels may be normal in patients with early myositis. In such cases, elevated lactate dehydrogenase and aminotransferase levels may represent early clues to the presence of muscle involvement [6, 7]. Magnetic resonance imaging (MRI) has become the method of choice for muscle assessment in pediatric and adult patients with suspected DM [15]. As the most sensitive modality available, MRI can detect mild muscle involvement in patients with no weakness or muscle enzyme alterations [16].
Most patients with ADM do not progress to clinically evident myositis. In three case series of patients with clinical ADM (a group that includes amyopathic and hypomyopathic subtypes) who received no systemic therapy, two of 28 adult patients (7 %) and none of 20 juvenile patients progressed to symptomatic muscle disease over a mean followup period of 5 years [4, 14, 17]. However, other authors have documented a higher incidence of calcinosis cutis in the subset of juvenile DM patients with skin manifestations preceding muscle involvement by more than 1 year, and calcinosis cutis developed in at least one pediatric patient in the aforementioned series [17]. Further study of the long-term outcomes of clinically ADM with and without systemic therapy are needed in order to establish guidelines for patient management.
References
1. Pearson CM. Polymyositis and dermatomyositis. In: McCarty DJ, ed. Arthritis and Allied Conditions: A Textbook of Rheumatology, 9th ed. Philadelphia: Lea & Febiger;1979:7422. Sontheimer RD. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis siné myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol 2002;46:626
3. Caproni M, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol 2002;138:23
4. El-Azhary RA, et al. Amyopathic dermatomyositis: retrospective review of 37 cases. J Am Acad Dermatol 2002;46:560
5. Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol 1998;39:899
6. Euwer RL, Sontheimer RD. Amyopathic dermatomyositis: a review. J Invest Dermatol 1993;100:124S
7. Wargula JC. Update on juvenile dermatomyositis: new advances in understanding its etiopathogenesis. Curr Opin Rheumatol 2003;15:595
8. Peloro TM, et al. Juvenile dermatomyositis: a retrospective review of a 30-year experience. J Am Acad Dermatol 2001;45:28
9. Ghali FE. Gingival telangiectasias: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol 1999;135:1370
10. Piantandida MD, et al. Infrapatellar hypertichosis: an unusual cutaneous manifestation of juvenile dermatomyositis. Pediatr Dermatol 2002;19:132
11. Tse S, et al. The arthritis of inflammatory childhood myositis syndromes. J Rheumatol 2001;28:192
12. Tezak Z, et al. Gene expression profile in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol 2002;168:4154.
13. Fedczyna T, et al. Expression of TNFalfa by muscle fibers in biopsies from children with untreated juvenile dermatomyositis: association with the TNFalfa-308 A allele. Clin Immunol 2001;100:236 14. Plamondon S, Dent PB. Juvenile amyopathic dermatomyositis: results of a case finding descriptive survey. J Rheumatol 2000;27:2031
15. Park JH, Olsen NJ. Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies. Curr Rheumatol Rep 2001;3:334
16. Maillard SM, et al. Quantitative assessment of MRI T2 relaxation time of the thigh muscles in juvenile dermatomyositis. Rheumatology 2004;43:603
17. Cosnes A, et al. Dermatomyositis without muscle weakness: long-term follow-up of 12 patients without systemic corticosteroids. Arch Dermatol 1995;131:1381
© 2005 Dermatology Online Journal