Alkaptonuria
Published Web Location
https://doi.org/10.5070/D37rt3x92bMain Content
Alkaptonuria
Molly Yancovitz MD, Robert Anolik MD, Miriam Keltz Pomeranz MD
Dermatology Online Journal 16 (11): 6
Department of Dermatology, New York University, New York, New York Abstract
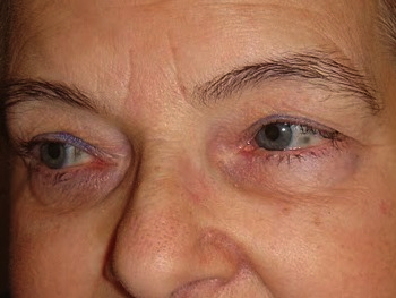
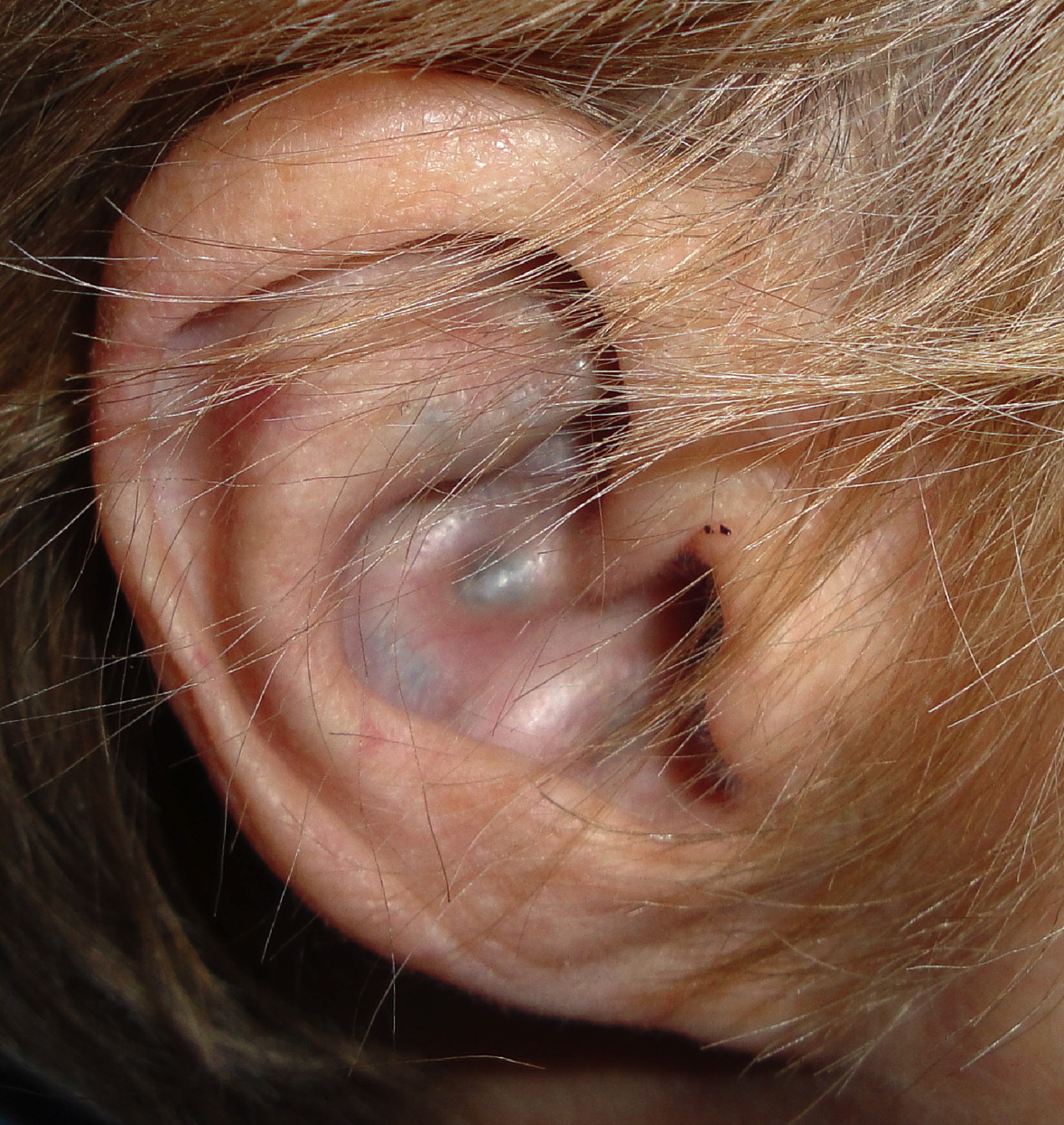
A 69-year-old woman presented with a 30-year history of lower back and large joint pain of the hips and shoulders. On examination blue-grey, pigmented macules were present over the cartilaginous portions of the ears and on the sclera. Past medical history included aortic stenosis. Urine homogentisic acid level was elevated, which is diagnostic for alkaptonuria. Alkaptonuria is an autosomal recessive disorder that results in deficiency of homogentisic acid oxidase and in the accumulation of homogentisic acid in connective tissue. Disease can result in blue-grey pigmentation of the cartilage, sclerae, face, and hands as well as severe arthropathy and cardiac valve disease. Treatment is limited at this time. Promising early reports of the use of nitisinone have prompted ongoing trials of this therapeutic agent.
History
 |  |
| Figure 1 | Figure 2 |
|---|
The patient presented to the Rheumatology Clinic at Bellevue Hospital Center for evaluation of a greater than 30-year history of arthritis, which started in her lower back and progressed to involve her hips and shoulders. She noted that her back and joint pain occurred intermittently throughout the day and often was worse with activity. At that time, she was noted to have a blue hue to her sclera and ear cartilage. Her medical history included left total hip arthroplasty, chronic obstructive pulmonary disease, osteoporosis, depression, aortic atherosclerosis, aortic stenosis, toxic multinodular goiter, and vulvar lichen sclerosus. Her medications included risendronate, albuterol, aspirin, inhaled tiotropium, inhaled fluticasone/salmerterol, ezetamibe-simvastatin, diclofenac, omeprazole, calcium, and vitamin D. Family history disclosed a brother and sister with arthritis and similar pigmentary changes and a grandfather with arthritis.
Physical examination
Blue-grey pigmented macules were present on the cartilaginous portions of the ears and on the sclera.
Laboratory data
Complete blood count, comprehensive metabolic panel, rheumatoid factor, erythrocyte sedimentation rate, C-reactive protein, uric acid, creatine kinase, and urinalysis were normal. Urine homogentisic acid level was elevated at 9,753 mmol/mol creatinine and on repeat sampling was 13,935 mmol/mol creatinine (controls <10). Antinuclear antibody was 1:320, with a nucleolar patten. Radiographic studies showed severe disc space narrowing at all levels of the cervical, thoracic, and lumbar spine, near complete ankylosis across the discovertebral joints at L1/2 through L4/5, and severe sclerosis at the sacroiliac joins and symphysis pubis.
Histopathology
None.
Comment
Alkaptonuria is an autosomal recessive disorder that results from deficient activity of homogentisic acid dioxygenase, which converts homogentisic acid to maleylacetoacetic acid in the tyrosine catabolic pathway. Mutations in the homogentisate 1,2-dioxygenase gene lead to a deficiency of homogentisic acid oxidase and results in the accumulation of homogentisic acid in connective tissues. The incidence is approximately 1:250,000 and is increased in certain countries including the Dominican Republic and Slovakia [1, 2, 3].
Elevated levels of homogentisic acid polymerize, which results in pigment deposition in connective tissue, the cause of ochronosis. Patients generally are asymptomatic in childhood and develop symptoms in the third-to-fourth decade of life. Cutaneous manifestations include blue or brown pigmentation, which initially affects the ears and sclerae. Pigmentation also can develop over the nose, cheeks, foreheard, extensor aspects of the hands, and axillary and inguinal areas. Perspiration can stain clothing. Patients may develop ochronotic arthritis, which frequently manifests as lower back pain and leads to kyphosis [4]. Patients may develop severe arthroparthy that involves the larger joints, such as the spine, hips, knees, and shoulders. Intervertebral disc calcification may be seen. Alkaptonuria also may cause cardiac valvular disease and coronary artery disease and lead to an increased incidence of myocardial infarcts. Alkptonuria patients also have a higher incidence of renal and prostatic stones [4].
Alkaptonuria is characterized by urine that initially appears normal and then turns brown or black after standing or after alkanization. The darkening is caused by the oxidation of homogentisic acid. Cloth diapers of affected infants may turn brown after washing with alkaline solutions. However, the diagnosis is most commonly made in adulthood as part of an evaluation for arthritis. Homogentisic acid levels are elevated in the blood, urine, and tissue specimens. Diagnosis is confirmed by urine homogentisic acid levels.
The differential diagnosis of the cutaneous findings of alkaptonuria includes exogenous ochronosis, argyria, chrysoderma, and drug-induced hyperpigmentation. Joint disease from alkaptonuria must be differentiated from ankylosing spondilitis, osteoarthritis, and other inflammatory arthropathies. Alkaptonuria is a rare cause of aortic valvular disease, which is more commonly caused by congenital, infectious, or degenerative processes.
Therapy for alkaptonuria is limited. The execretion of homogentisic acid is reduced with dietary restriction of tyrosine and phenylalanine, but little chemical effect has been demonstrated with this treatment [5]. Ascorbic acid has been given to inhibit the enzyme that catalyzes homogentisic acid oxidation but does not reduce homogentisic acid excretion and has yet to be shown to be efficacious [6]. Reports of the use of nitisinone, which blocks the accumulation of homogentisic acid, have shown decreased urinary excreation of homogentisic acid; some patients reported subjective improvement in joint pain. Larger clinical trials of this agent are underway [7, 8].
References
1. Goicoechea De Jorge E et al. Alkaptronuria in the Dominican Republic : identification of the founder AKU muation and further evidence of mutation hot spots in the HGO gene. J Med Genet 2002; 39: E40 [PubMed]2. Vilboux T, et al. Mutation spectrum of homogentisic acid oxidase (HGD) in alkaptornuria. Hum Mutat 2009; 30: 1611 [PubMed]
3. Sren, S, et al. Alkaptonuria in Slovakia: thirty-two years of research on phenotype and genotype. Mol Genet Metab 2002; 75: 353 [PubMed]
4. Phornphutkul C, et al. Natural history of alkaptonuria. N Engl J Med 2002; 327: 2111 [PubMed]
5. de Haas V, et al. The success of dietary protein restriction in alkaptonuria patients is age-dependent. J Inherit Metab Dis 1998; 21: 791 [PubMed]
6. Wolff JA, et al. Effects of ascorbic acid in alkptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res 1989; 26: 140 [PubMed]
7. Sunwanarat P, et al. Use of nitisinone in patients with alakptonuria. Metabolism 2005; 54: 719 [PubMed]
8. Long-Term Study the Nitisinone to Treat Alkaptonuria. [cited; Available from: http://clinicaltrials.gov/ct2/show/NCT00107783
© 2010 Dermatology Online Journal