Hypocomplementemic urticarial vasculitis in mixed connective tissue disease
Published Web Location
https://doi.org/10.5070/D37rd90671Main Content
Hypocomplementemic urticarial vasculitis in mixed connective tissue disease
Ana Maria Calistru MD1, Carmen Lisboa MD PhD1, Maria João Cruz MD1, Luis Delgado MD PhD2, Licínio Poças MD3, Filomena Azevedo MD1
Dermatology Online Journal 16 (12): 8
1. Department of Dermatology and Venereology, Hospital São João, Porto, Portugal2. Department of Immunology, Hospital São João, Faculty of Medicine, University of Porto, Portugal
3. Department of Rheumatology, Hospital São João, Porto, Portugal
Abstract
Urticarial vasculitis is characterized clinically by urticaria-like skin lesions and histologically by leukocytoclastic vasculitis. It may be idiopathic or associated with various conditions such as infections, hematologic disorders, drugs, and connective tissue diseases, primarily systemic lupus erythematosus; an association with mixed connective tissue disease (MCTD) has rarely been reported. We present a case of hypocomplementemic urticarial vasculitis in a patient with MCTD that responded to hydroxychloroquine after a period of corticosteroid dependence.
Introduction
Urticarial vasculitis is characterized by episodes of persistent urticarial skin lesions, with or without angioedema, and histological evidence of leukocytoclastic vasculitis. Patients can be divided into two groups, those with normal complement levels and those with hypocomplementemic urticarial vasculitis. The latter are more likely to have systemic compromise including arthralgia, progressive lung disease, ocular inflammation, and renal involvement [1]. Urticarial vasculitis is frequently idiopathic but may be associated with serum sickness, drugs, physical urticaria, Schnitzler syndrome, infections, hematologic disorders, and connective tissue diseases, primarily systemic lupus erythematosus. Nevertheless, the association with mixed connective tissue disease (MCTD) has been rarely reported. We present a case of hypocomplementemic urticarial vasculitis in a patient with MCTD that responded to prednisone followed by maintenance treatment with hydroxychloroquine.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
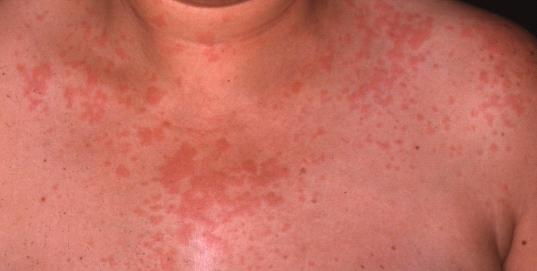
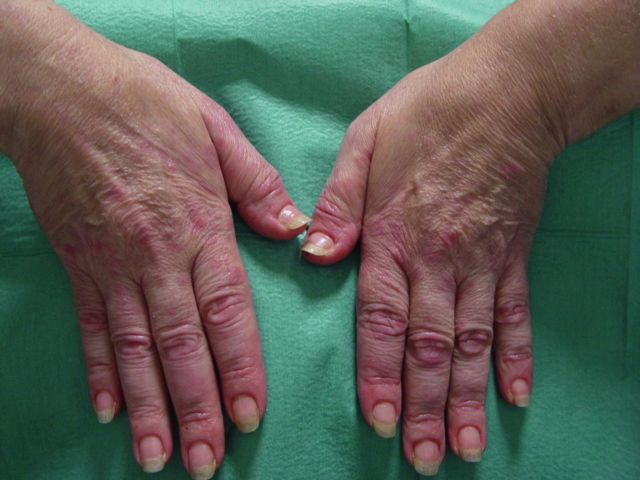
| Figure 1. Urticarial papules and plaques on the trunk. Figure 2. Edema of the hands. | |
 |
| Figure 3 |
|---|
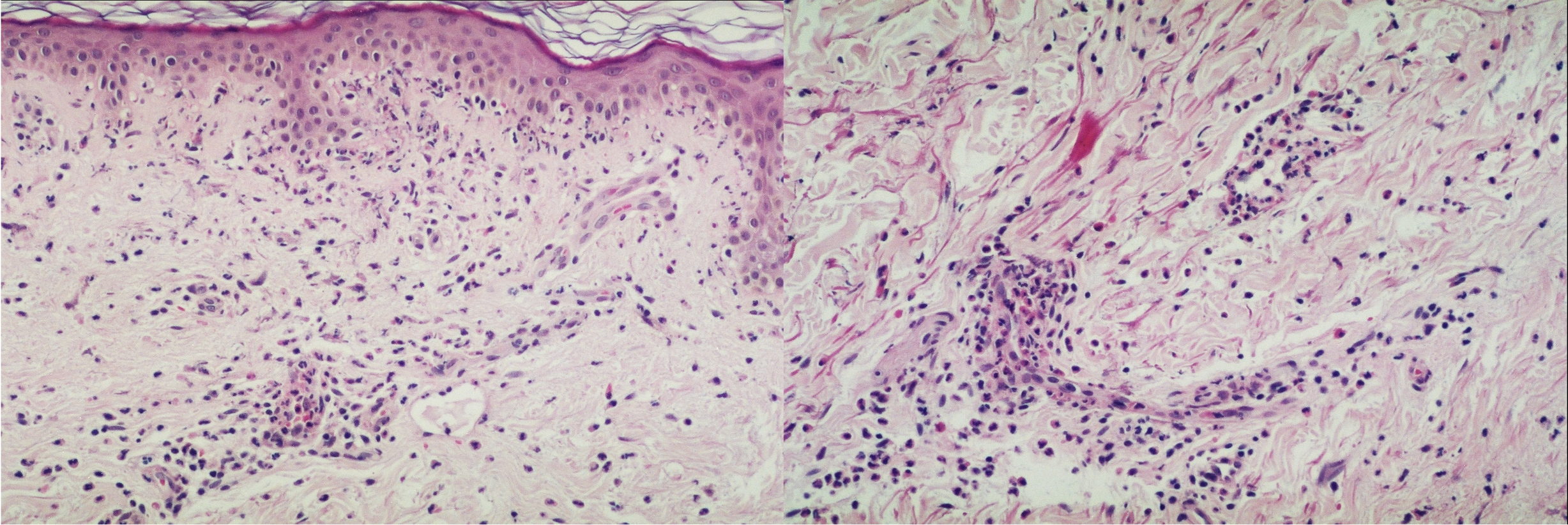
| Figure 3. Small vessel vasculitis in the dermis (left), with neutrophilic infiltrate, presence of leukocytoclasis and red blood cells extravasation (inset, right), consistent with leukocytoclastic vasculitis (H&E). |
A 56-year-old woman presented to the Dermatology department with a 3-week history of recurrent generalized urticarial papules and plaques, unresponsive to antihistaminic treatment (Figure 1). The eruption had a purpuric quality,and was associated with mild pruritus. Individual lesions persisted beyond 48 hours. She also had a 5-year history of edema of the hands (Figure 2), Raynaud phenomenon, and hand and foot arthralgias for which she was receiving intermittent treatment with indomethacin. The patient denied difficulty in swallowing, dyspnea, myalgia, muscle weakness or other symptoms. She was otherwise healthy and no other drug intake was mentioned. The histopathologic examination of a skin biopsy specimen revealed damage of the small vessels in the dermis with neutrophilic infiltrate, and presence of leukocytoclasis and red blood cells extravasation, consistent with leukocytoclastic vasculitis (Figure 3).
Complete blood cell count, erythrocyte sedimentation rate, chemistry panel including aldolase and creatine kinase, C-reactive protein, and urinalysis were within normal range. The antibody serologies of hepatitis B, hepatitis C, HIV, cytomegalovirus, and Epstein Barr virus were negative. Rheumatoid factor, anti-double stranded DNA, anti-cardiolipin, anti-centromere, and antineutrophil cytoplasmic antibodies were all negative. The protein electrophoresis showed an elevated level of gamma globulins (25%; normal range 10.5-19.5%) with increased immunoglobulin G (1890 mg/dl; normal range 650-1500 mg/dl). The C1q level (4 mg/dl; normal range, 18 – 45 mg/dl), C3c level (27 mg/dl; normal range, 83-177 mg/dl), C4 level (7 mg/dl; normal range, 12-36 mg/dl) were decreased, whereas the circulating immune complex (CIC) level was markedly increased (100 μg/ml; normal < 4.4 μg/ml). An elevated titer of antinuclear antibodies (ANA) was detected (>1/1000; speckled). There was a high titer of autoantibodies against U1-ribonucleoprotein (U1RNP), as shown by the immunoblot test. The other anti-extractable nuclear antigen (ENA) antibodies (anti-Jo1, anti-SCL70, anti-Sm, anti-Ro, anti-La) were negative. Nailfold capillaroscopy and chest radiography were unremarkable. HLA-DR4 typing was negative.
The patient’s urticarial skin lesions with histological evidence of leukocytoclastic vasculitis and decreased complement levels were diagnosed as hypocomplementemic urticarial vasculitis. The diagnosis of MCTD was established because of the presence of Raynaud phenomenon, edema of the hands, and arthralgias (synovitis) associated with a high titer of anti-U1RNP autoantibodies.
Treatment with oral prednisone was started, initially 0.5 mg/kg/day and then 1 mg/kg/day; resolution of the skin lesions and improvement in arthralgias was achieved. Laboratory improvement included an increase of the C1q level (from 4 to 14 mg/dl), and decrease of the CIC level (from >100 to 22 μg/ml). The attempts at tapering the corticosteroid dose were accompanied by the recurrence of the urticarial papules and plaques, therefore, after 4 months, hydrohychloroquine 400 mg/day was added. The prednisone was slowly tapered and, at one year follow-up, the patient was free of symptoms while receiving prednisone 10 mg every other day and hydrohychloroquine 400 mg/day. During follow-up, high titers of ANA (>1/1000, speckled) and anti-U1RNP antibodies were still detected.
Discussion
The association between urticarial vasculitis and MCTD has been rarely reported. There are two reviews describing 3 patients with MCTD among 68 cases of urticarial vasculitis [2], and one patient among 27 [3].
The initial description of MCTD as a distinct connective tissue disease was made by Sharp and colleagues in 1972 [4]. Since then, the concept has been controversial, but now is evident that MCTD is a distinct entity. The presence of high titers of autoantibodies to U1RNP influences the expression of connective tissue disease in ways that are relevant to prognosis and treatment [5]. Mixed connective tissue disease is characterized by overlapping features of lupus erythematosus, systemic sclerosis, polymyositis / dermatomyositis, and rheumatoid arthritis. There are various published criteria for the diagnosis of MCTD. The Alarcon-Segovia criteria include high titers of antibodies against U1RNP as well as the presence of at least 3 of 5 of the following clinical features: edema of the hands, synovitis, myositis, Raynaud phenomenon, and acrosclerosis [6]. This critera set has a sensitivity of greater than 90 percent, a specificity of greater than 98 percent [7]. It was evaluated as one of the best classification criteria to define MCTD [8]. Other clinical features are pulmonary interstitial fibrosis, pulmonary hypertension, esophageal reflux or dysmotility, small-vessel vasculitis with palpable purpura, leg ulcers, alopecia, facial erythema, and periungueal telangiectasias. Renal involvement is rare and usually mild. Anti-U1RNP antibodies are detectable in 25-47 percent of SLE patients, but high titers of anti-U1RNP are diagnostic of MCTD [9]. There is a strong association of MCTD with HLA-DR4, which is found in 52 percent of the patients [10]. The prognosis varies from a benign course to severe progressive disease. In approximately one-third of patients the clinical symptoms go into long-term remission and the anti-RNP antibodies disappear. One-third of patients have a severe and progressive disease course [11]. The primary disease-related cause of death is pulmonary involvement [12].
Systemic corticosteroid is the most effective treatment of hypocomplementemic urticarial vasculitis. Management also includes indomethacin and antihistamines for symptomatic relief, dapsone, colchicine, and hydroxychloroquine. For refractory cases azathioprine, methotrexate, cyclophosphamide, and rituximab have also been used [1]. However, therapy of HUV involves management of the underlying cause, when identified. Because no controlled studies have been performed to guide the therapy in MCTD, the conventional therapies used in other rheumatic conditions are employed. Corticosteroids, antimalarials, methotrexate, cytotoxics, and vasodilators have all been used [13]. Hydroxychloroquine inhibits chemotaxis of eosinophils and locomotion of neutrophils; in addition, it impairs complement-dependent antigen-antibody reactions. Our patient responded favorably to oral steroids (1 mg/kg/day) but the frequent relapses with tapering, in the setting of mild systemic involvement, made hydroxychloroquine an appropriate choice.
This case illustrates the relevance of searching for underlying disease in patients with urticarial vasculitis, especially when it occurs with hypocomplementemia.
The diagnostic criteria for MCTD are based on a specific serologic marker (U1-RNP), the association with HLA-DR4, the mild renal involvement, and the predisposition to pulmonary disease. These seem to delimit a clinically distinct and stable condition, but the longitudinal follow-up of MCTD patients must take into account the possible evolution to another entity in the connective tissue spectrum, such as systemic sclerosis, systemic lupus erythematosus, and dermatomyositis.
References
1. Saigal K, Valencia IC, Cohen J, Kerdel FA. Hypocomplementemic urticarial vasculitis with angioedema, a rare presentation of systemic lupus erythematosus: Rapid response to rituximab. J Am Acad Dermatol. 2003 Nov; 49(5 Suppl):S283-5. [PubMed]2. Dincy CVP, George R, Jacob M, Mathai E, Pulimood S, Eapen EP. Clinicopathologic profile of normocomplementemic and hypocomplementemic urticarial vasculitis: a study from South India. J Eur Acad Dermatol Venereol. 2008 Jul; 22(7):789-94. [PubMed]
3. Asherson RA, D'Cruz D, Stephens CJ, McKee PH, Hughes GR. Urticarial vasculitis in a connective tissue disease clinic: patterns, presentations, and treatment. Semin Arthritis Rheum. 1991 Apr; 20(5):285-96. [PubMed]
4. Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed Connective Tissue Disease - an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972 Feb; 52(2):148-59. [PubMed]
5. Maddison PJ. Mixed connective tissue disease: overlap syndromes. Baillieres Best Pract Res Clin Rheumatol. 2000 Mar; 14(1):111-24. [PubMed]
6. Alarcon-Segovia D, Villareal M. Classification and diagnostic criteria for mixed connective tissue disease, in: Kasukawa R, Sharps GC (Eds), Mixed Connective Tissue Disease and Anti-Nuclear Antibodies, Elsevier, Amsterdam, 1987, pp. 33-40
7. Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989 Mar; 16(3):328-34. [PubMed]
8. Amigues JM, Cantagrel A, Abbal M, Mazieres B. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. Autoimmunity Group of the Hospitals of Toulouse. J Rheumatol. 1996 Dec; 23(12):2055-62. [PubMed]
9. Migliorini P, Baldini C, Rocchi V, Bombardieri S. Anti-Sm and anti-RNP antibodies. Autoimmunity. 2005 Feb; 38(1):47-54. [PubMed]
10. Ruuska P, Hämeenkorpi R, Forsberg S, Julkunen H, Mäkitalo R, Ilonen J, and Tiilikainen A. Differences in HLA antigens between patients with mixed connective tissue disease and systemic lupus erythematosus. Ann Rheum Dis. 1992 Jan; 51(1): 52-55. [PubMed]
11. Lundberg IE. The prognosis of mixed connective tissue disease. Rheum Dis Clin North Am. 2005 Aug;31(3):535-47, vii-viii. [PubMed]
12. Hoffman RW, Maldonado ME. Immune pathogenesis of Mixed Connective Tissue Disease: a short analytical review. Clin Immunol. 2008 Jul; 128(1):8-17. [PubMed]
13. Kim P, Grossman JM. Treatment of mixed connective tissue disease. Rheum Dis Clin North Am. 2005 Aug; 31(3):549-65,viii. [PubMed]
© 2010 Dermatology Online Journal