Acral lymphomatoid papulosis associated with poikilodermatous mycosis fungoides

Main Content
Acral lymphomatoid papulosis associated with poikilodermatous mycosis fungoides
Julie Magorien1,4 BS, Joseph D Hillman2,4 MD, Lauren C Pinter-Brown3,4 MD, Jonathan Said2,4 MD, Melvin W Chiu1,4,5 MD, MPH
Dermatology Online Journal 19 (2): 1
1. Division of Dermatology, Department of Medicine, David Geffen School of Medicine at the University of California, Los Angeles,
California2. Division of Dermatopathology, Department of Pathology, David Geffen School of Medicine at the University of California, Los Angeles, California
3. Division of Hematology-Oncology, Department of Medicine, David Geffen School of Medicine at the University of California, Los Angeles, California
4. David Geffen School of Medicine at the University of California, Los Angeles
5. Dermatology Service, West Los Angeles Veterans Affairs Medical Center, Los Angeles, California
Abstract
We present a case of a 51-year-old man with concomitant acral lymphomatoid papulosis and poikilodermatous mycosis fungoides.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
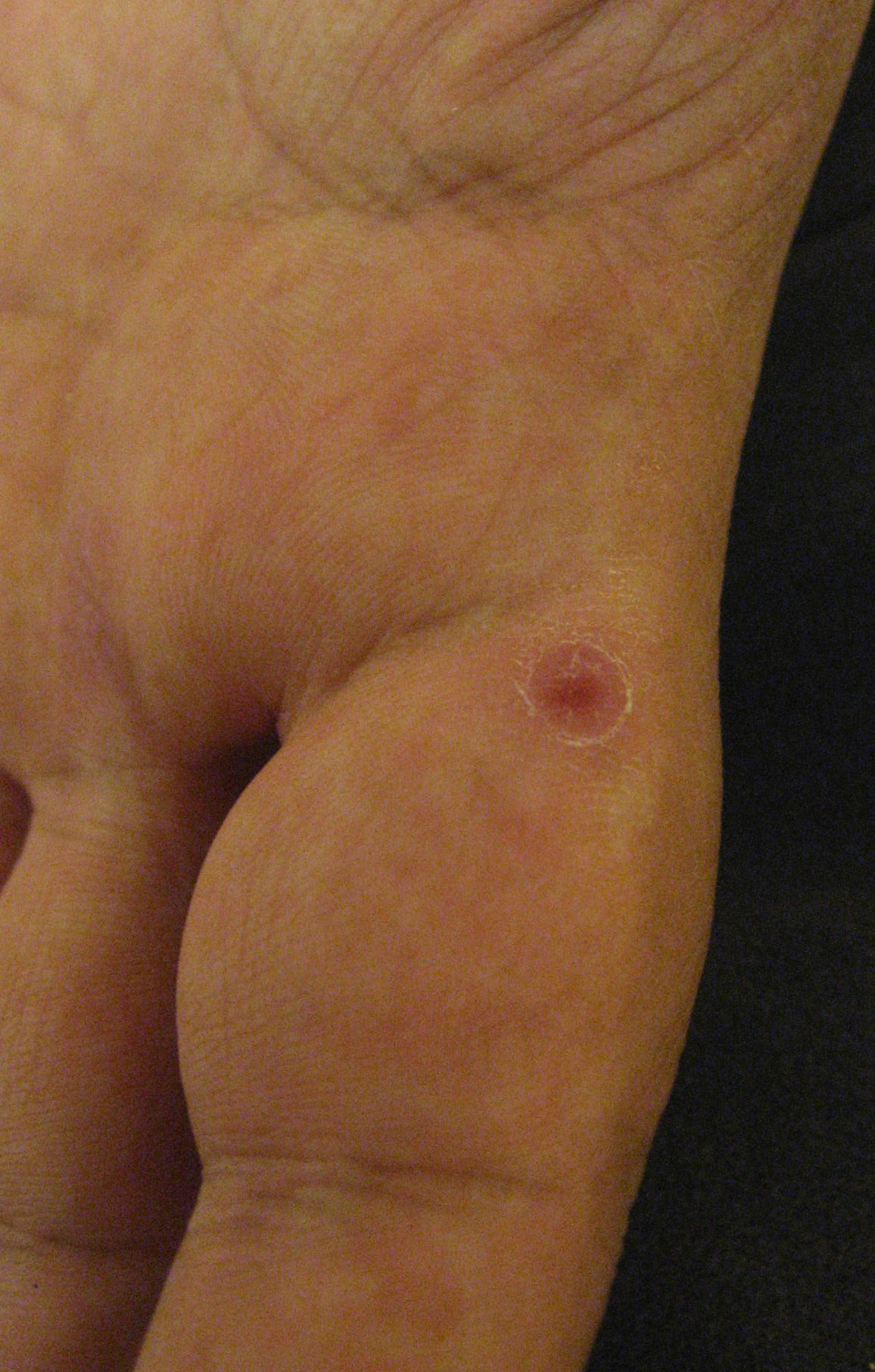
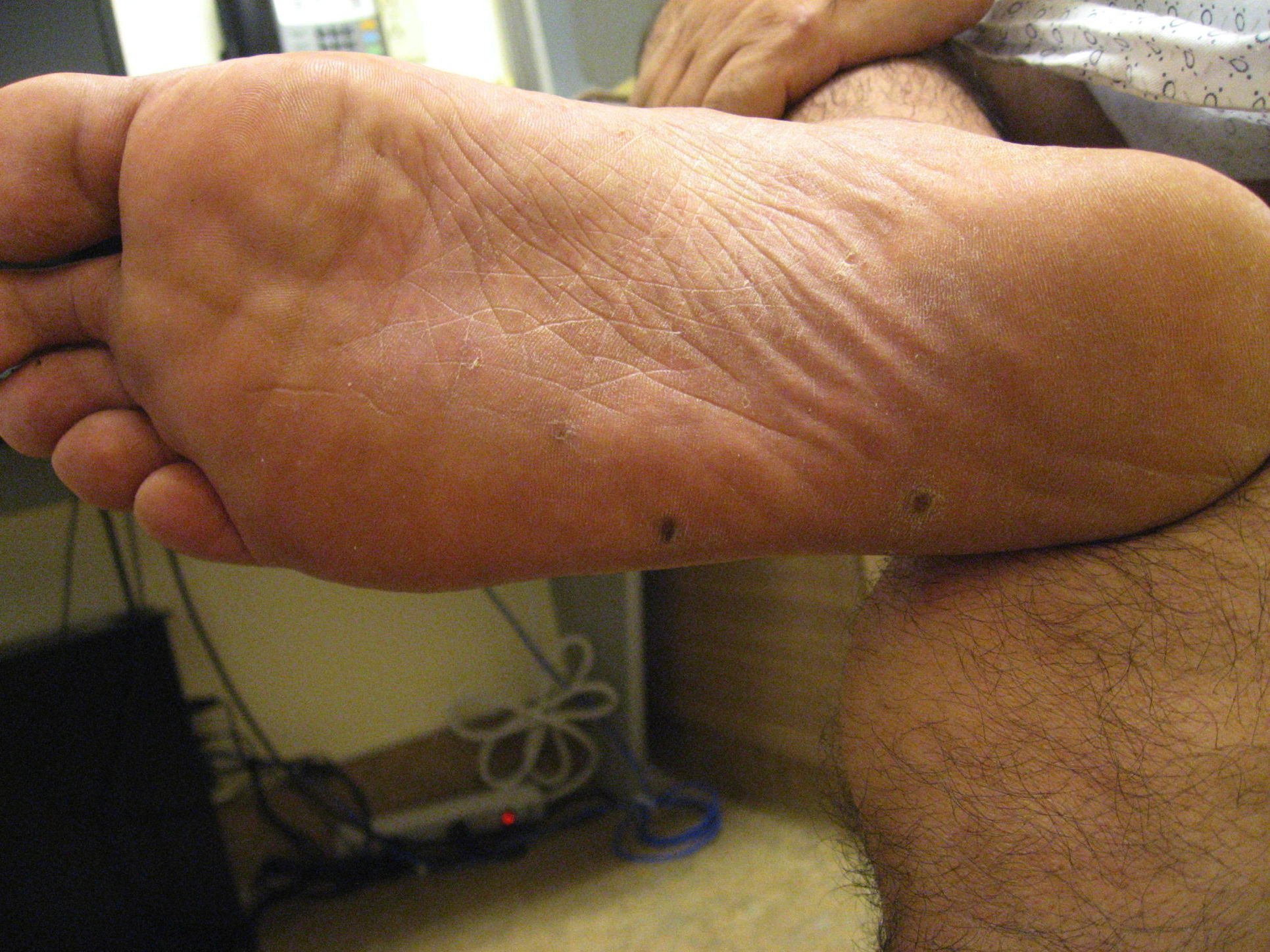
| Figure 1. A representative pink, crusted macule with scale on the volar finger Figure 2. Crusted papules on the plantar foot. | |
A 51-year-old man with a several year history of “chronic eczema” presented with a painful 1 cm red papule on the plantar surface of his left foot. The lesion resolved after treatment with cephalexin prescribed for suspicion of cellulitis. Five months later, similar red, crusted papules appeared bilaterally on the patient’s palms and fingers (Figure 1) as well as the plantar aspects of his feet (Figure 2). The lesions were characterized as “burning” but not overtly painful. After treatment with intramuscular triamcinolone injections for a presumed diagnosis of eczema, the lesions involuted, but then similar papulonodular lesions, which would spontaneously improve and then recur, appeared at different locations along the fingers and acral surface of the hands.
 |  |
| Figure 3 | Figure 4 |
|---|---|
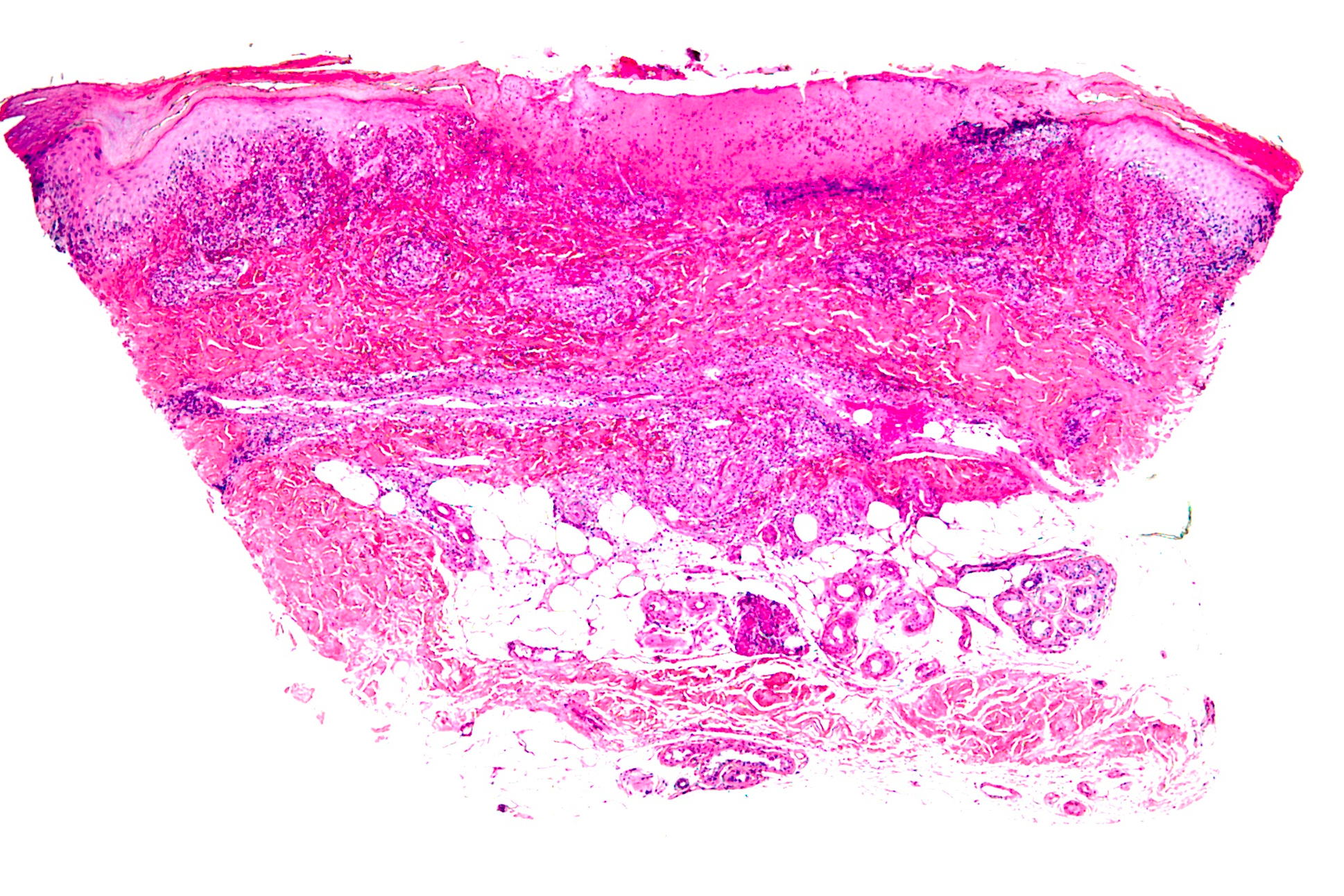
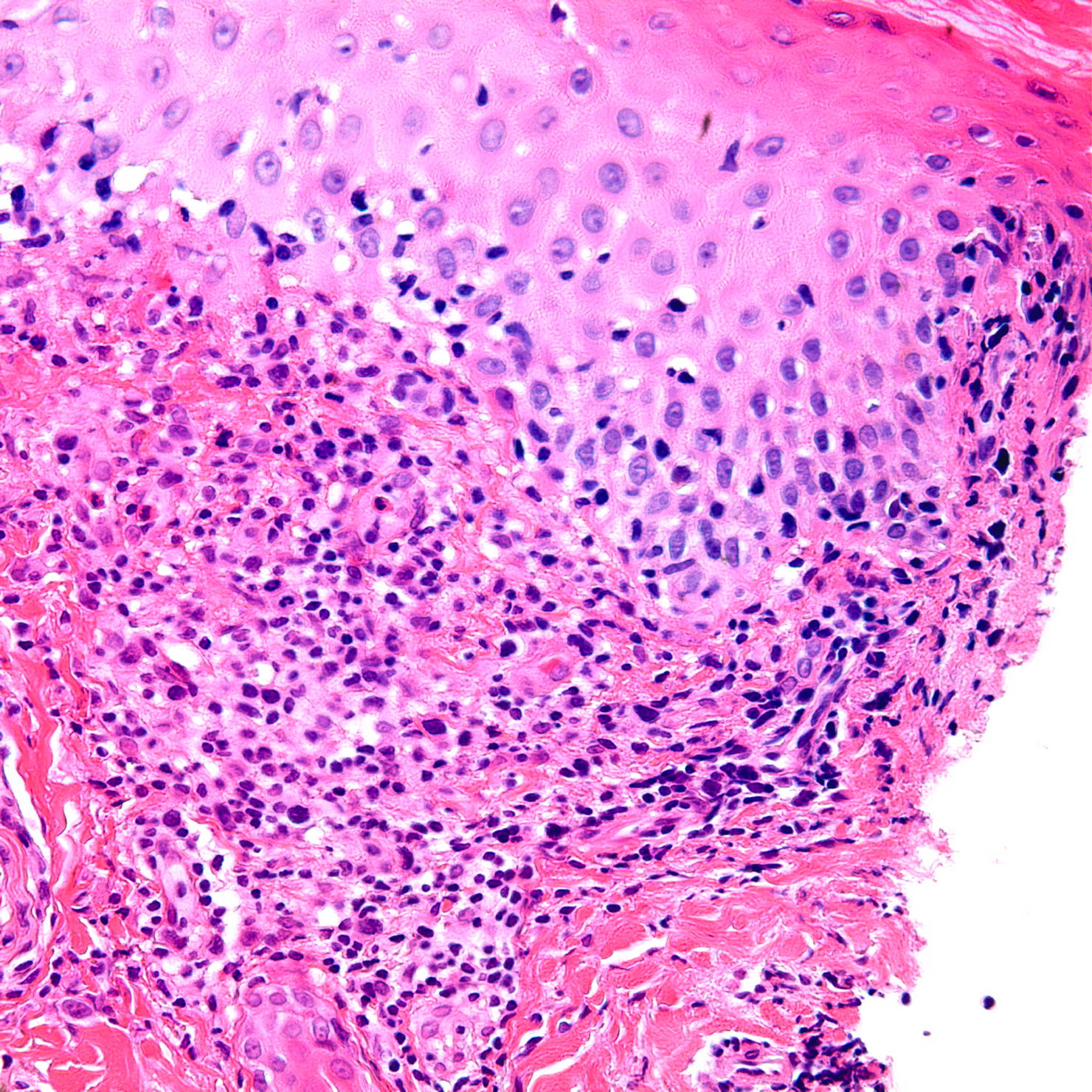
| Figure 3. An ulcer and a vaguely wedge-shaped inflammatory infiltrate in the dermis. (H&E x4) Figure 4. Large lymphoid cells with hyperchromatic, enlarged nuclei. (H&E x40) | |
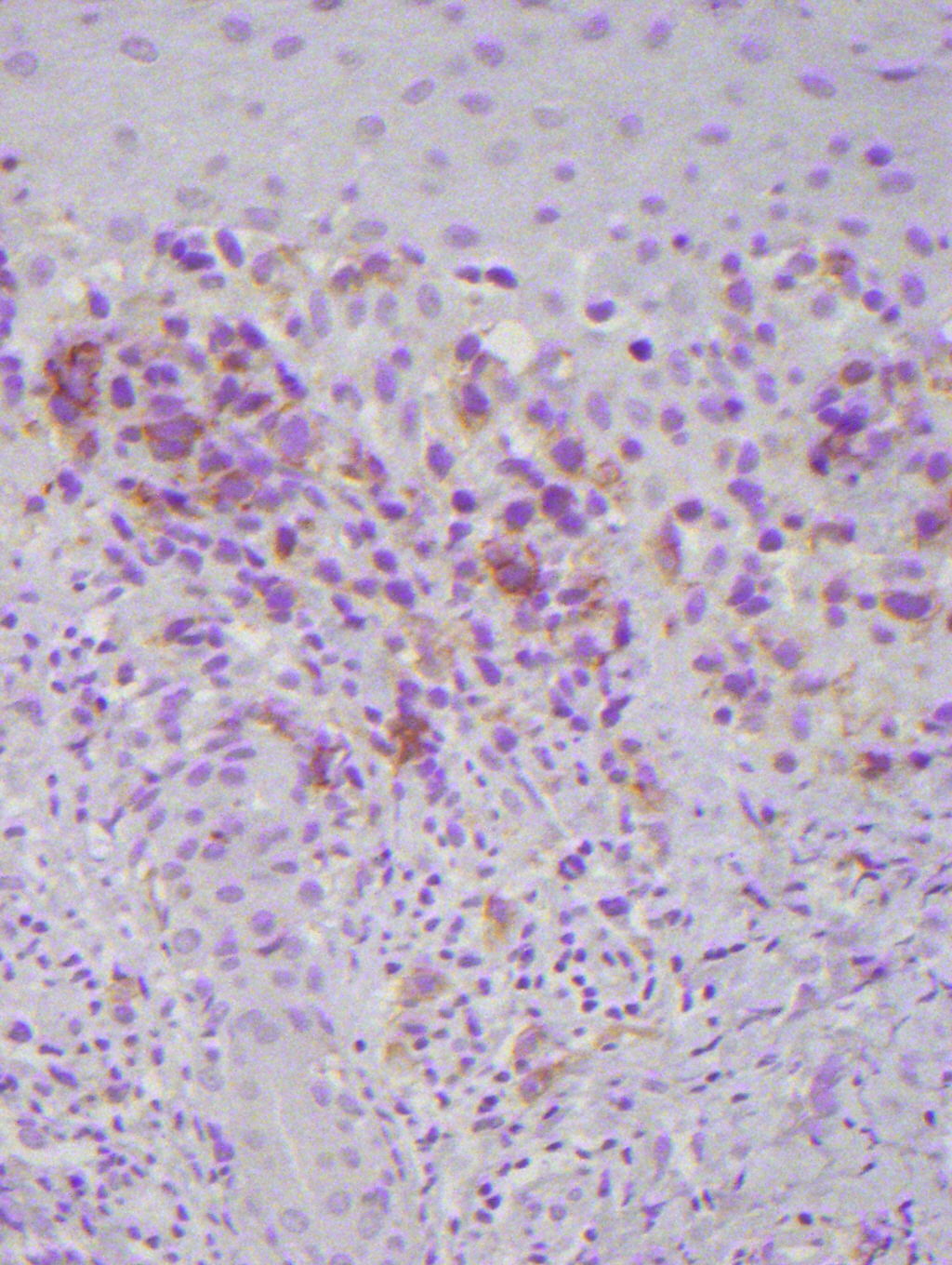
Histopathology from a representative lesion revealed an atypical perivascular lymphoid infiltrate of the dermis with epidermotropism and ulceration (Figures 3 and 4). The lymphoid cells were characterized by both small and large cells with highly irregular contours and open chromatin. Mitotic figures were present. The atypical lymphoid cells were positive for CD3, CD5, and CD30 by immunohistochemistry (Figure 5). The atypical cells were negative for ALK, CD20, CD43, CD56, and Langerin. Given the combination of his clinical and histologic findings, the patient was diagnosed with acral lymphomatoid papulosis (LyP).
 |  |
| Figure 5 | Figure 6 |
|---|---|
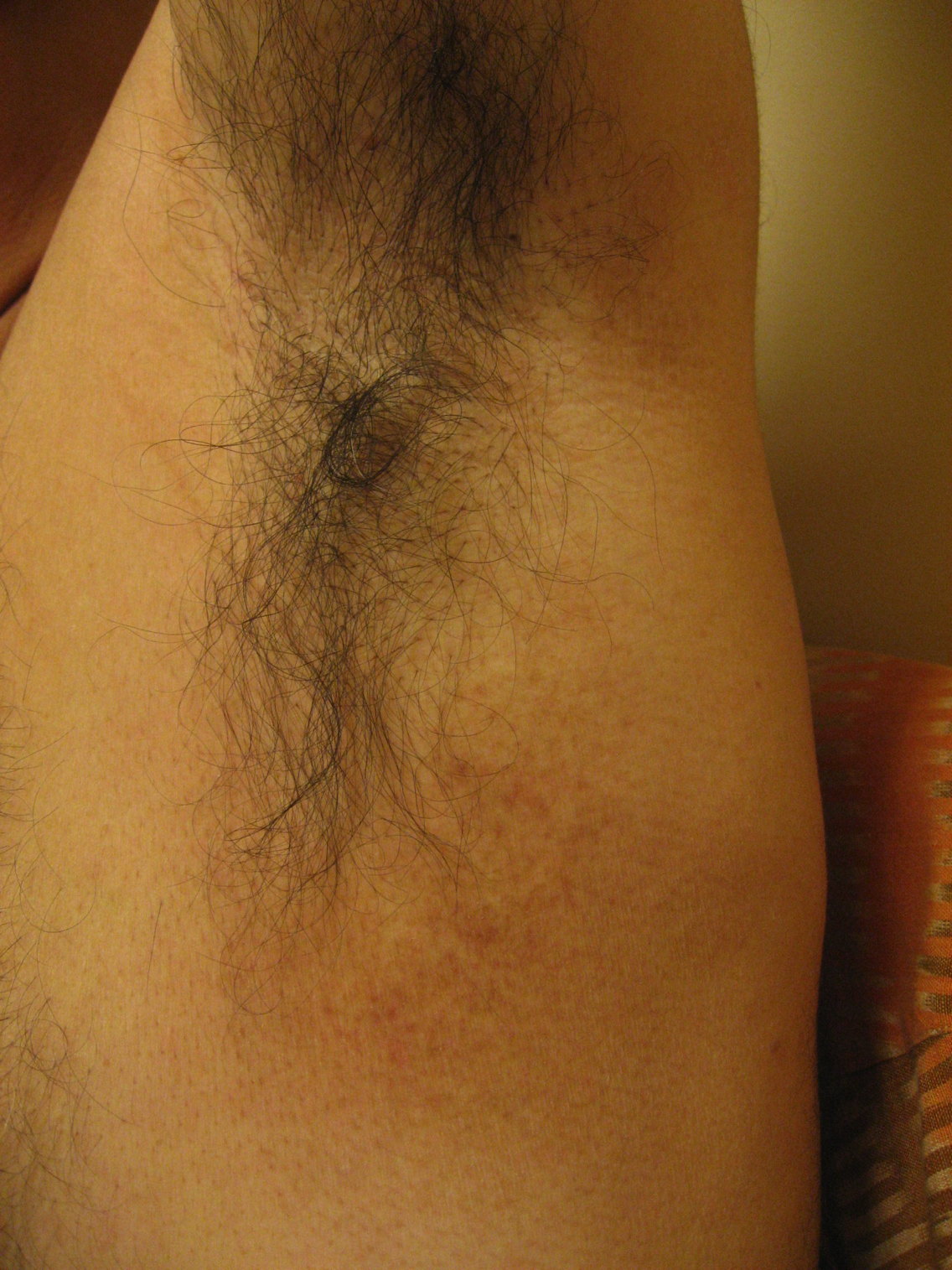
| Figure 5. CD30 positive lymphoid cells (immunoperoxidase x40) Figure 6. Poikilodermatous patch in the axilla | |
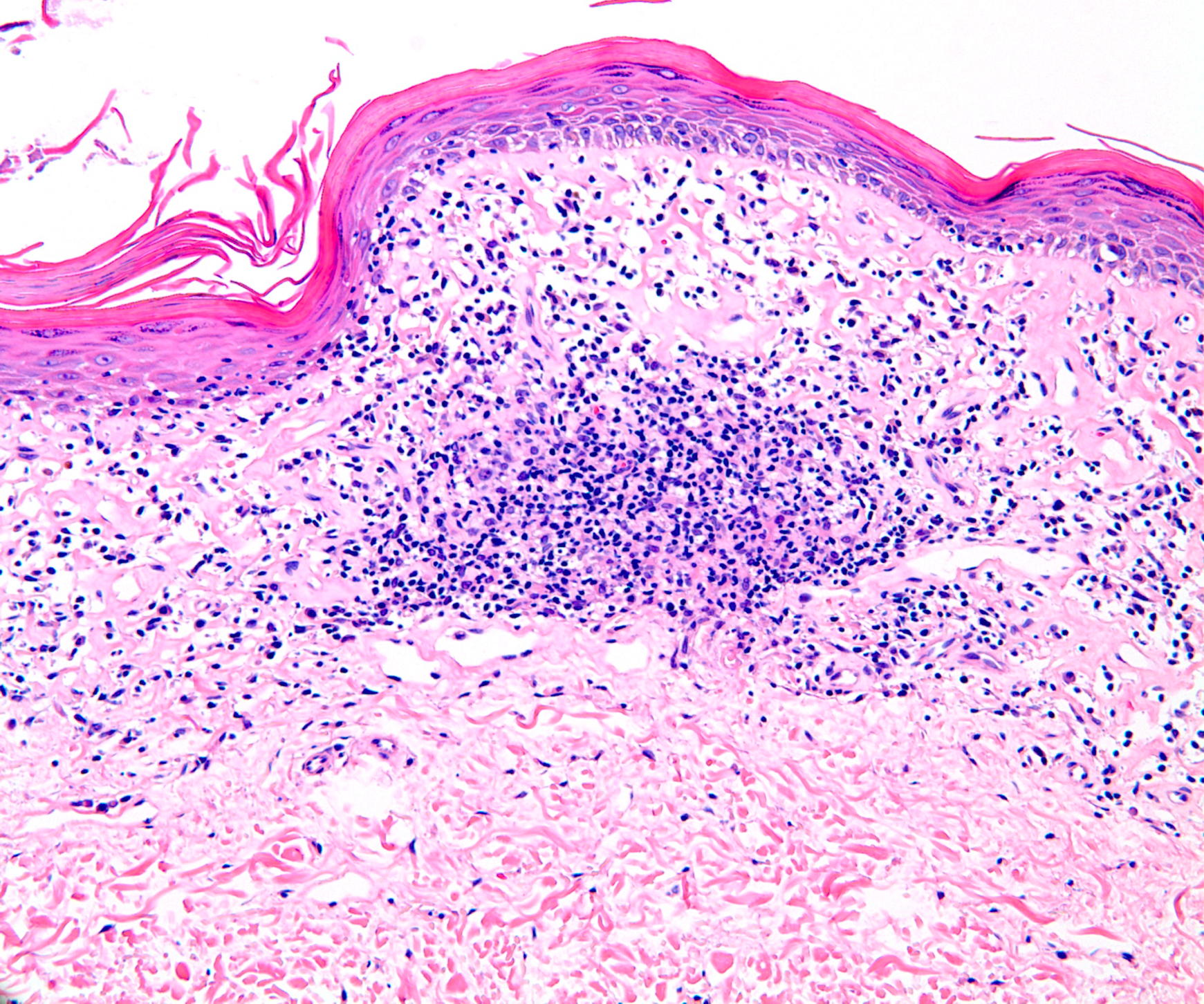
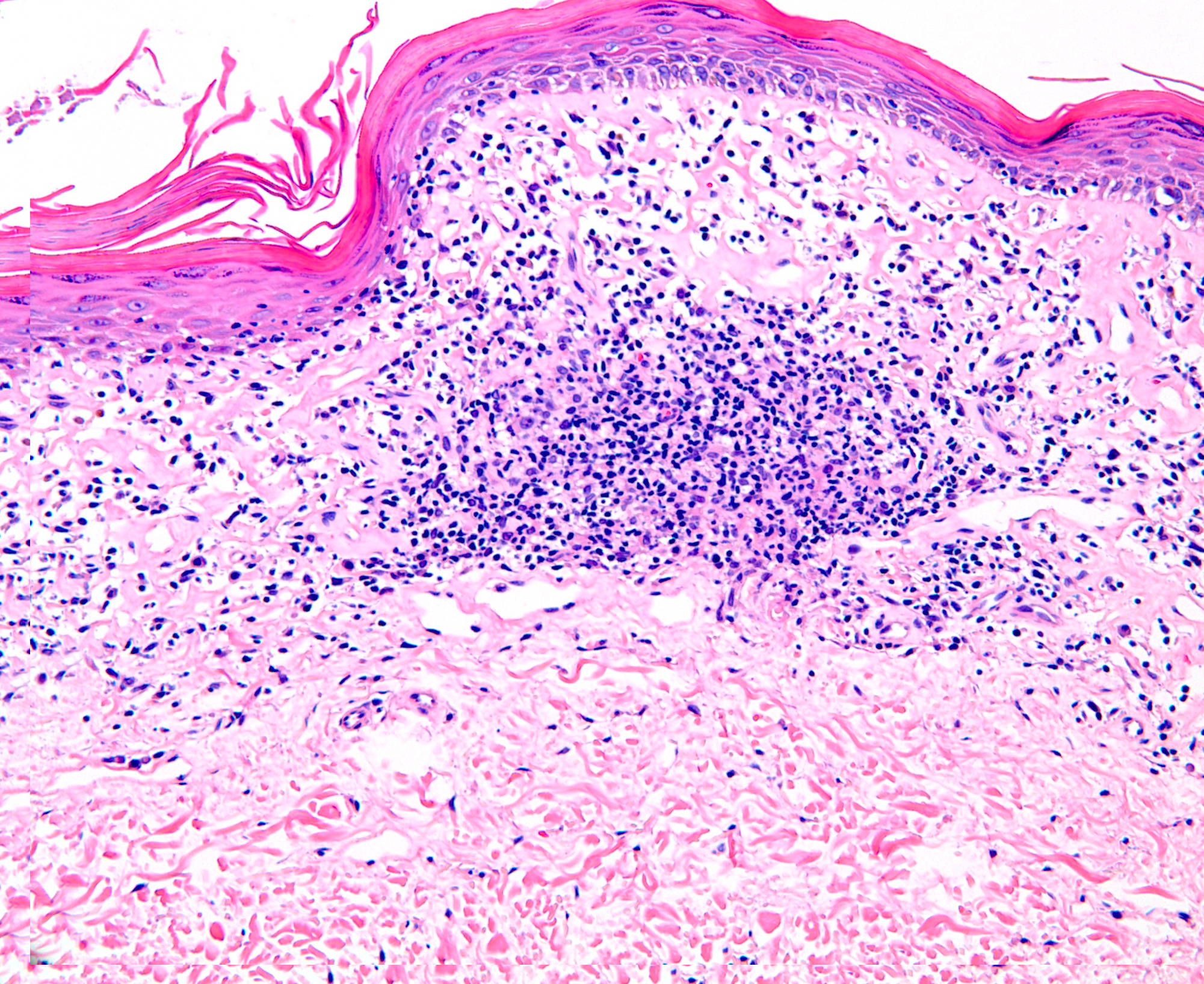
The patient also was found to have asymptomatic poikilodermatous patches on his groin and axilla (Figure 6). These patches had been present and persistent for over 20 years and were representative of his “chronic eczema.” Biopsies of these areas revealed atypical lymphocytic infiltrates within the papillary dermis (Figures 7 and 8). The infiltrates were comprised predominately of T cells, which lined up at the dermal-epidermal junction. The T cells were marked by irregular nuclear contours. Immunohistochemistry analysis showed atypical cells positive for CD3, CD4, CD2, and CD5. Notably, these cells showed loss of CD7 and had a clonal gamma T cell receptor gene rearrangement by polymerase chain reaction (PCR) analysis. This histological picture suggested mycosis fungoides (MF). Our patient began treatment with narrowband UVB (NBUVB) phototherapy with improvement in both his LyP and his MF.
 |  |
| Figure 7 | Figure 8 |
|---|---|
| Figure 7. A band-like lymphocyte-predominant inflammatory infiltrate and wiry bundles of collagen in the superficial dermis.
(H&E x20) Figure 8. Atypical lymphocytes in a linear array at the dermal-epidermal junction and atypical lymphocytes present within the epidermis above the dermal-epidermal junction. (H&E x40) | |
Discussion
This case demonstrates acral LyP coexisting with poikilodermatous MF of the axilla and groin. This is an unusual presentation of LyP because LyP is characteristically confined to the trunk and limbs, but is rarely located on the hands [1, 2, 3, 4]. LyP is a primary cutaneous CD30+ lymphoproliferative disorder (LPD). Papulonodular lesions develop, ulcerate, and then spontaneously heal in 3-12 weeks [5]. Patients will typically have multiple lesions of the trunk and limbs at varying stages of healing. LyP generally affects adults with a slight predilection for men (median age 45, male-to-female ratio, 1.5:1) [5, 6]. Although the histologic hallmark of LyP is anaplastic, aggressive-appearing CD30+ lymphoid cells, in fact, patients with LyP have a favorable prognosis. LyP can be further categorized into 1 of 4 types based on histological findings [5]. Type A LyP, the most common type, has a wedge-shaped infiltrate composed of large atypical CD30+ lymphoid cells among abundant dermal inflammatory cells such as neutrophils, histiocytes, eosinophils, and small lymphocytes [5]. Type B LyP is marked by small epidermotropic atypical lymphocytes with cerebriform nuclei, bearing a resemblance to the histologic pattern seen in MF. This patient’s LyP histology is most consistent with type A/B overlap. Type C LyP features large atypical CD30+ cells like type A LyP, with the distinction that the cells are aggregated in sheets or nodules and contain fewer inflammatory cells. Type D LyP is marked by CD8+ CD30+ lymphoid cells similar to the histologic picture seen in primary cutaneous CD8+ epidermotropic T cell lymphoma [7]. Recently, an angioinvasive CD30+ CD8+ variant of LyP that simulates aggressive angiocentric T cell lymphomas has been described. This has been classified as type E [8].
Primary cutaneous CD30+ LPDs are considered a spectrum of disease consisting of LyP and primary cutaneous anaplastic large cell lymphoma (PCALCL) [5, 7]. The two CD30+ LPDs have several overlapping features. Often, histology alone cannot differentiate LyP from PCALCL, but the clinical exam and course can help delineate the CD30+ LPDs. Unlike the crops of papulonodular lesions seen in LyP, patients with PCALCL typically have solitary tumors, grouped nodules, or plaques with ulceration [5, 7]. Seldom, a patient can present with multifocal lesions. Similar to the papulonodules of LyP, the tumors of PCALCL can spontaneously regress without treatment. Histologically, PCALCL is marked by sheets of large atypical CD30+ cells. Moreover, at least 75 percent of the atypical tumor cells are CD30+ in PCALCL [7].
Patients with CD30+ LPDs are reported to be at increased risk for other neoplasms such as MF, Hodgkin disease, and Non-Hodgkin lymphoma [5, 9]. Several reports have found that 10 to 20 percent of cases of LyP are associated with other lymphomas [5, 6, 10]. LyP is associated specifically with MF in 7 percent to 39 percent of cases [6, 10, 11]. In a study of 49 patients with poikilodermatous MF, 18 percent had coexistent LyP [12]. The authors concluded that LyP was seen more often in poikilodermatous MF than in other variants of cutaneous T-cell lymphoma (4%) [12]. The association between LyP and lymphoma is explained by a primary T cell clonal expansion manifesting as more than one lymphoproliferative disease in a patient. This theory is supported by PCR analysis, which found that patients with coexisting LyP and MF can have identical T cells clones from both dermatoses [11, 13]. Specifically, PCR analysis revealed identical T cell receptor-gamma gene sequences from MF and LyP specimens from the same patient [10]. It is thought that the original clonal expansion can acquire further mutations and ultimately develop into a second distinct malignancy.
The differential diagnosis of CD30+ LPDs also includes CD30+ large cell transformation (LCT) of MF. Per Herrmann et al, there are 3 presentations of LCT including a new solitary nodule within a classic MF patch or plaque, a sudden eruption of pink scattered papules or nodules without spontaneous involution, or a new or enlarging tumor [14]. The histopathologic criteria for LCT include large cells, four times the size of a lymphocyte, comprising greater than 25 percent of the infiltrate or large cells forming microscopic nodules [15]. Distinguishing LCT from CD30+ LPDs like LyP and PCALCL can be challenging. If fewer than 75 percent of the large cells stain positive for CD30 and the clinical presentation is consistent with LCT, one can be fairly certain that the diagnosis is LCT [16]. However, if greater than 75 percent of the large cells stain positive for CD30, then histology alone cannot distinguish LCT from CD30+ lymphoproliferative diseases [16]. The clinical findings and course must be accounted for. Clinically, LCT can occur in all stages of MF, but is more likely to occur in patients with advanced stages of MF. Arulogun found that 1.4 percent of patients with stage I MF went on to develop LCT whereas 50 percent of patients with stage IV MF developed LCT [17].
Our patient was diagnosed with coexistent LyP and MF rather than MF with LCT given the patient’s clinical presentation and histological findings from cutaneous biopsy. First, the patient’s pink papules on his palms and plantar aspects of his feet would spontaneously resolve, which is consistent with LyP and not LCT. The papules did not form within the patient’s poikilodermatous patches of his axilla and groin and he did not develop any new or enlarging tumors, which are both consistent with LCT. Furthermore, the patient had stage 1A MF, and LCT is more likely to occur in patients with advanced stages of MF [17]. Finally, the histological findings from cutaneous biopsy of the patient’s papules were consistent with Type A LyP.
A workup for newly diagnosed LyP can begin with a thorough history and physical, complete blood count with differential, chemistry panel, and lactate dehydrogenase [7]. The history should include a detailed dermatologic history, personal and family history of lymphoma, and a history of B symptoms [7]. Generally, this is sufficient if there is no clinical or laboratory evidence of extracutaneous disease [7]. In cases of LyP, in which extracutaneous disease is suspected, the patient should undergo imaging studies and lymph node biopsy, if applicable [7].
The prognosis for LyP is favorable. Only about 4 percent of patients with LyP will progress to systemic lymphoma [6]. One study found that of 118 patients with LyP, only 2 patients died from an associated lymphoma [6]. Interestingly, when MF coincides with LyP, the prognosis is more favorable than MF alone [11, 12, 13, 18]. In one study of 21 patients with coexistent LyP and MF, all of the patients were alive up to 21 years follow up [10]. Similarly, a combined series of 19 cases of coexistent LyP and MF, all patients were alive with one patient progressing to nodal disease [19]. Another study of 15 patients with LyP and MF, all patients were alive with over a decade since diagnosis of MF [11]. MF alone has a 10-year survival rate of 80 percent [11].
LyP is an incurable disease and, to date, there are no treatments proven to significantly alter the course of disease or prevent development of additional lymphoprolferative disease processes. Moreover, it is challenging to determine the efficacy of treatment because LyP lesions spontaneously heal. An argument can be made to initially abstain from treatment altogether given the favorable prognosis of LyP [7]. However, various treatment modalities are available to mitigate lesions especially in cases where the lesions are profuse or painful. The most common treatments are topical corticosteroids, oral methotrexate, photochemotherapy, and topical chemotherapy [7]. Unfortunately, the cure rates are relatively low with recurrence rates reaching 40 percent with treatment [7]. Other studies report successful treatment of LyP with bexarotene, NBUVB, or 308-nm excimer laser [20, 21]. Conclusive randomized controlled trials are still needed to study the most effective treatments for LyP.
The prognosis of poikilodermatous MF is also favorable. In Abbott’s case series, 88 percent had early stage (earlier than Stage IIA) disease, and all patients remained alive after a median of 8 years of follow-up [12]. Most patients respond well to phototherapy (PUVA, UVB, or NBUVB). Other treatments include superficial radiotherapy, methotrexate, or systemic bexarotene [12].
Our case highlights two important issues. One, it is imperative to rule out LCT in a patient with suspected coexistent LyP and MF, becausethe management and prognosis of LCT differs greatly from that of LyP. The second issue is the importance of thorough workup given the well-documented association between LyP and lymphoproliferative disease. Effective management and care of these patients necessitate an interdisciplinary approach among dermatology, pathology, and oncology.
References
1. Kagaya M, Kondo S, Kamada A, et al., Localized lymphomatoid papulosis, Dermatology 2002;204(1):72-74. [PubMed]2. Yancovitz M, Walters RF, Kamino H, et al. Acral lymphomatoid papulosis. J Am Acad Dermatol. 2010 Mar;62(3):530-1. [PubMed]
3. Deroo-Berger MC, Skowron F, Ronger S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205(1):60-2. [PubMed]
4. Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998 Mar-Apr;15(2):146-7. [PubMed]
5. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005 May 15;105(10):3768-85. [PubMed]
6. Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000 Jun 15;95(12):3653-61. [PubMed]
7. Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011 Oct 13;118(15):4024-35. [PubMed]
8. Kempf W, Kazakov DV, Scharer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol 2012 Sep 28. [Epub ahead of print] [PubMed]
9. Wang HH, Myers T, Lach LJ, et al. Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis. Cancer 1999;86:1240-1245. [PubMed]
10. Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003 Oct;49(4):620-3. [PubMed]
11. Basarab T, Fraser-Andrews EA, Orchard G, et al, Lymphomatoid papulosis in association with mycosis fungoides: A study of 15 cases. Br J Dermatol 1998;139(4):630-8. [PubMed]
12. Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011 Aug;65(2):313-9. [PubMed]
13. Gallardo F, Costa C, Bellosillo B, et al. Lymphomatoid papulosis associated with mycosis fungoides: clinicopathological and molecular studies of 12 cases. Acta Derm Venereol. 2004;84(6):463-8. [PubMed]
14. Herrmann JL, Hughey LC. Recognizing large-cell transformation of mycosis fungoides. J Am Acad Dermatol. 2012 Jan 18. Epub ahead of print [PubMed]
15. Salhany KE, Cousar JB, Greer JP, et al. Transformation of cutaneous T cell lymphoma into large cell lymphoma. A clinicopatholofic and immunologic study. Am J Pathol 1988; 132:265-77. [PubMed]
16. Vergier B, de Muret A, Beylot-Barry M, et al. Transformation of mycosis fungoides: clinicopathological and prognostic features of 45 cases. French Study Group of Cutaneious Lymphomas. Blood. 2000 Apr 1;95(7):2212-8. [PubMed]
17. Arulogun SO, Prince HM, Ng J, et al. Long-term outcomes of patients with advanced-stage cutaneous T-cell lymphoma and large cell transformation. Blood. 2008 Oct;112(8):3082-7. [PubMed]
18. Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010 Nov 1;28(31):4730-9. Epub 2010 Sep 20. [PubMed]
19. Beljaards RC, Willemze R. The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br J Dermatol. 1992 Jun;126(6):596-602. [PubMed]
20. Krathen RA, Ward S, Duvic M. Bexarotene is a new treatment option for lymphomatoid papulosis. Dermatology. 2003;206(2):142-7. [PubMed]
21. Kontos AP, Kerr HA, Malick F, et al. 308-nm excimer laser for the treatment of lymphomatoid papulosis and stage IA mycosis fungoides. Photodermatol Photoimmunol Photomed. 2006 Jun;22(3):168-71. [PubMed]
© 2013 Dermatology Online Journal