Three cousins with chronic foot ulcers resulting from late-onset hereditary sensory and autonomic neuropathies type 2 (HSAN2)
Published Web Location
https://doi.org/10.5070/D37k21s656Main Content
Three cousins with chronic foot ulcers resulting from late-onset hereditary sensory and autonomic neuropathies type 2 (HSAN2)
Shahin Aghaei MD 1, and Kambiz Pakmanesh MD2
Dermatology Online Journal 12 (2): 5
Department of Dermatology1, and Department of Rehabilitation2, Jahrom Medical School, Jahrom, Iran. shahinaghaei@yahoo.comAbstract
The hereditary sensory and autonomic neuropathies (HSAN) are a group of rare disorders characterized by prominent sensory and autonomic neuropathy without motor involvement. We report three male cousins with chronic foot ulcers, all were affected with late-onset HSAN type 2 (HSAN2). In view of the history of consanguinity and male sex, X-linked recessive transmission was likely in our patients. According to the authors' knowledge this is the first report of HSAN2 from Iran.
Introduction
The hereditary sensory and autonomic neuropathies (HSAN) are a group of rare disorders characterized by prominent sensory and autonomic neuropathy without motor involvement [1]. They reflect failure of development or degeneration of subpopulations of peripheral sensory and autonomic neurons. These disorders are classified into five main groups based on inheritance, clinical features, and the population of sensory neurons affected. Impaired pain appreciation results in mutilating acropathy with skin ulceration and fissuring, long bone fractures, Charcot joints, and digit amputation. The precise symptoms and signs and the nerve conduction abnormalities of each type are determined by the subpopulation of sensory neurons predominantly affected [2, 3]. We report on three male cousins affected with late-onset HSAN type 2 (HSAN2).
Clinical synopses
Three male cousins ages 17, 15, and 14 years old, presented with history of insensitivity to pain and temperature and multiple painless ulcerations over various parts of the upper and lower extremities since 6, 7, and 5 years of age, respectively. They were born of consanguineous marriage in three Muslim families. All were born at full term by normal vaginal delivery at a hospital and had uneventful antenatal, intranatal and postnatal periods. There was no history of recurrent episodes of fever. The bladder and bowel habits were normal. The immunization history was complete. The course of the disease was slowly progressive, the eldest cousins being most affected. Both parents of each patient were phenotypically normal. There was no history of abortion or stillbirth. There was a positive family history of such illness in paternal side.
 |  |
| Figure 1 | Figure 2 |
|---|---|
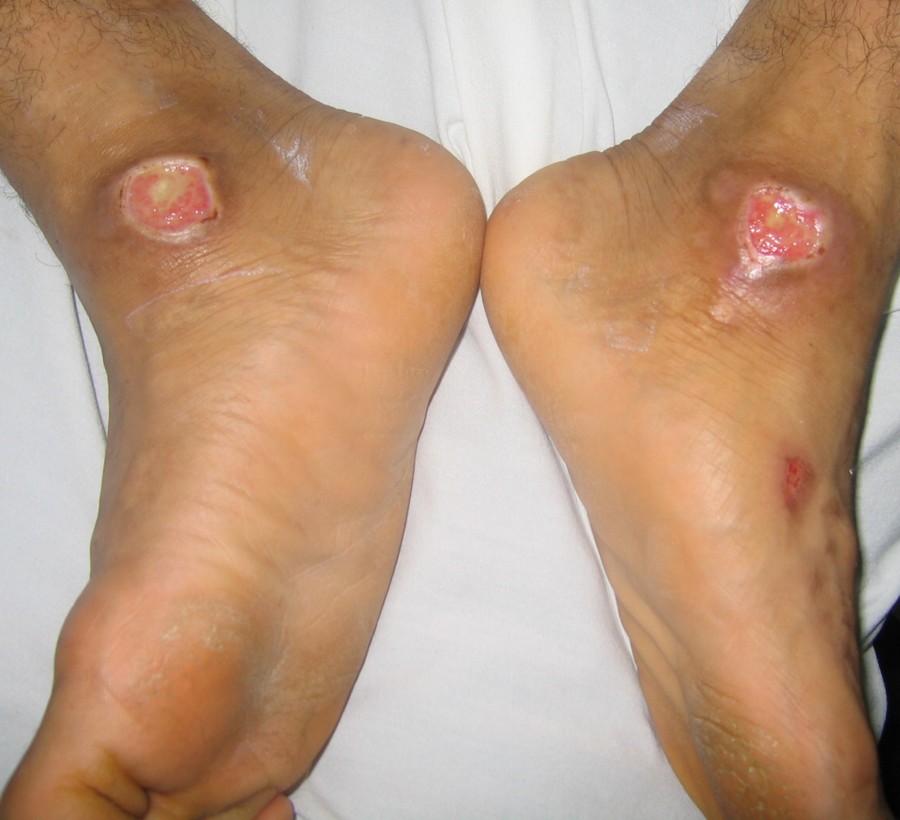
| Case 1 with chronic ulcers on backs of the hands and feet and medial malleoli. Amputated toes are seen. | |
 |  |
| Figure 3 | Figure 4 |
|---|---|
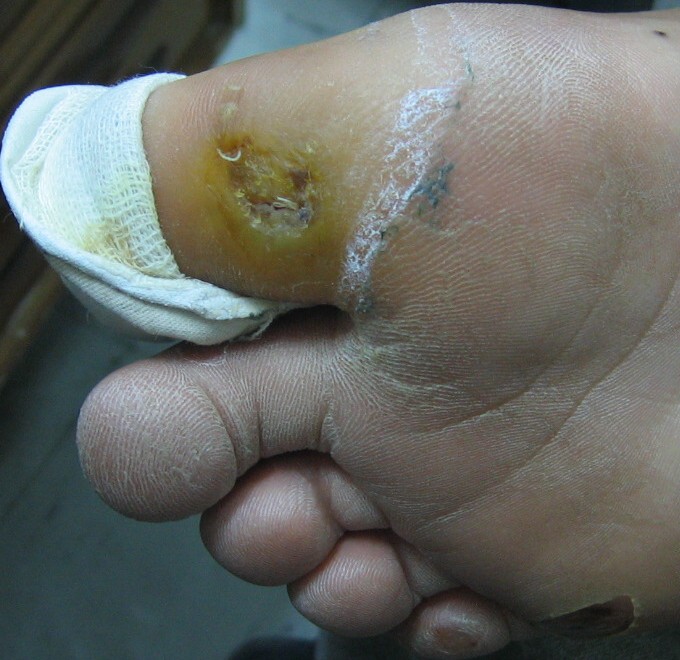
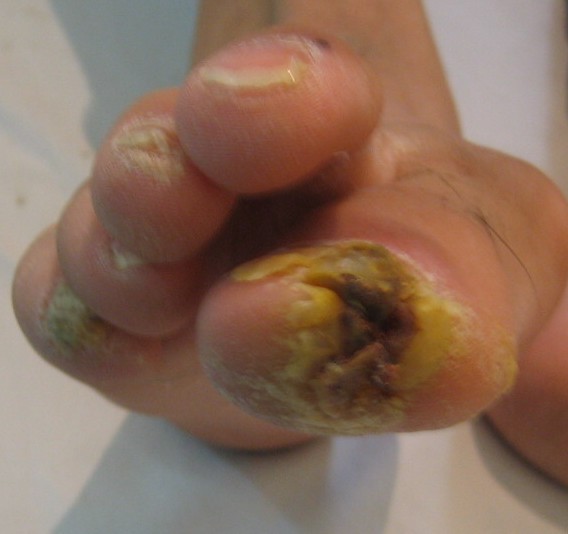
| Case 2 and 3, respectively, with a chronic hyperkeratotic ulcer on the big toe. | |
 |
| Figure 5 |
|---|
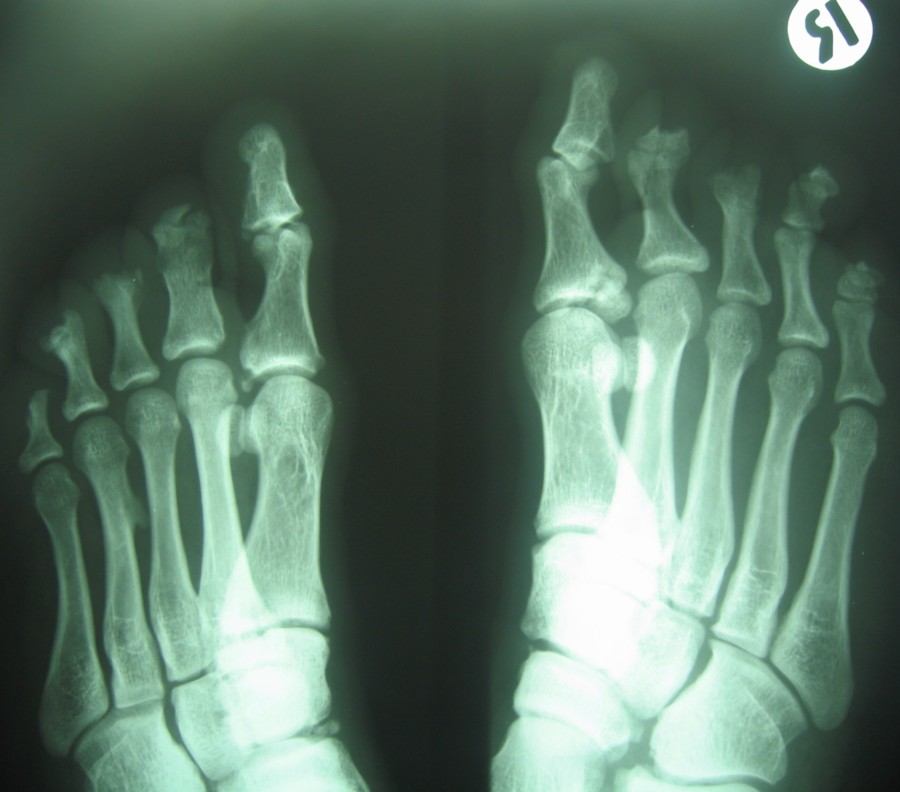
| X-ray view of the case 1. |
Examination revealed generalized loss of all modalities of sensation, more marked on the distal parts of the limbs. Multiple ulcerations were present on backs of the hands and feet in case 1 (Figs. 1 and 2) and only on right big toe in cases 2 and 3 (Figs. 3 and 4). No ulcers occurred on the lips or alae of the nose. Only one of the patients (case 1) showed mutilating acropathy in the form of complete or partial loss of toes and foot deformities (Fig. 5). Other features included anhidrosis over distal parts of limbs, and callosities on the palms and soles. There was no evidence of corneal opacities or bowing of legs. There was no sign of peripheral nerve thickening. Except total areflexia (superficial and deep), motor functions were well preserved. Fundus examination and the remainder of the central nervous system and other systemic examinations including blood pressure were normal. Routine hematological investigations were within normal limits. Detailed biochemical assessment was done in all. Serum calcium, phosphorus, blood urea and creatinine were within normal limits. Based on the above features, a provisional clinical diagnosis of HSAN2 was made and electrophysiological studies were performed in all the patients to confirm the diagnosis. Nerve biopsies were not done because of the lack of the pathological nerve examination capabilities in our institute. In all the patients conventional electrophysiological studies were done under standard conditions with control of limb temperature. Sensory nerve action potentials were unobtainable throughout. All compound muscle action potentials (CMAPs) amplitudes were within normal ranges, and motor nerve conduction velocities were low normal. Needle electromyographic examinations were essentially normal.
The patients were managed conservatively. Infected ulcers were treated with antibiotics and antiseptic dressings. Proper foot and skin care was ensured and adequate counseling was done to prevent trauma and improve personal hygiene to prevent infections.
Discussion
A group of rare disorders described as hereditary sensory and autonomic neuropathies (HSAN) can be classified into five different types [3]. The HSAN1 is perhaps the most common of these hereditary disorders. It is autosomal dominant with symptom onset in the second through fourth decades with sensory loss and subsequent tissue injury. This is helpful in distinguishing HSAN1 from other types of HSAN, which typically have an early onset. Insensitivity to pain is more prominent than decreased touch sensation. Sural nerve biopsy shows loss of unmyelinated fibers more than the myelinated ones [3, 4]. The HSAN2 is recessively inherited and begins at birth or in infancy. There is generalized pansensory loss. Autonomic disturbances include bladder dysfunction, impotence, and anhidrosis of hands and feet. Motor function is preserved but tendon reflexes are usually lost. Association of retinitis pigmentosa, motor weakness, and neurotrophic keratitis with sensory neuropathy has been described [1, 2, 3, 5]. Sensory nerve action potentials are absent and there is loss of myelinated fibers in sural nerve biopsy [1]. The HSAN types 3, 4, and 5 are also autosomal recessive, and type 3 is associated with prominent autonomic involvement [1, 6]. Nerve biopsy shows reduced number of unmyelinated fibres. HSAN4 is associated with episodic fevers, failure to thrive, postural hypotension, anhidrosis, self-mutilation, and mental retardation. Unlike type 3, sympathetic skin responses are unobtainable in type 4 [7]. Clinical manifestations of HSAN5 include mutilating acropathy and bilateral neurotrophic keratitis. Motor functions and tendon reflexes are normal. Sural nerve biopsy shows selective reduction of smaller myelinated nerve fiber population [2].
In view of the history of consanguinity and male sex, X-linked recessive transmission was likely in our patients. Clinical features and nerve conduction velocity studies of all the patients correlate well with those of HSAN2 [1, 2, 8]. A similar form of HSAN may be inherited as X-linked recessive trait [9]. Our patients developed their disease in preschool age, unusual because HSAN type-II usually manifests at birth or in infancy [1, 2, 3, 5]. The usual autonomic disturbances in the form of bladder and bowel involvement and hypertension were absent in our cases, although all had distal anhidrosis. In this regard it is highlighted that anhidrosis is a prominent manifestation of HSAN4. Motor functions were well preserved. Association of spastic paraplegia with HSAN2 has been reported by Cavanagh et al. [10] although such feature was absent in our cases.
When faced with a child presenting with insensitivity to pain, impaired sweating, limb ulcers, and mutilation, HSAN should be considered in the differential diagnosis. HSAN can be differentiated from other diseases by the characteristic clinical and electrophysiological manifestations in accordance to its type. Lesch-Nyhan syndrome is an X-linked recessive disorder clinically characterized by lesions from self-mutilation. Hyperuricemia, a major diagnostic criterion for Lesch-Nyhan syndrome, is not found in HSAN [11]. Hereditary anhidrotic ectodermal dysplasia and Fabry disease (angiokeratoma corporis diffusum) are X-linked hereditary disorders having complete clinical expression only in males. Hereditary anhidrotic ectodermal dysplasia is characterized by typical facies, with scalp and eyebrow hypotrichosis, and major dental abnormalities; in contrast to HSAN, it is not accompanied by peripheral neurologic manifestations. Fabry disease is characterized by the presence of paroxismal pain and paresthesia, fever, transient proteinuria, and cutaneous angiokeratomas [11, 12].
Uniformity in the presence or absence of clinical features was seen in all except for unilateral involvement in cases 2 and 3. HSAN is a rare disorder in Iran. Initial delay in diagnosis can occur due to similarity of the mutilating acropathy with that of leprosy, a more common disease in this area. Hence the leprosy should always be excluded prior to diagnosis of this rare disorder. According to the authors' knowledge, this is the first report of HSAN2 from Iran.
All the described features may not be found in a single case of HSAN. Intragroup variations are present and are still being reported [13, 14]. The spectrum of clinical features, nerve conduction studies, inheritance patterns and nerve biopsies form the mainstay of diagnosis and subtyping. There are no medical therapies available to treat these neuropathies, other than prevention and treatment of mutilating skin and bone lesions. Prevention of trauma to the anesthetized parts and appropriate management of the sequels of autonomic neuropathy are the aims. Prognosis is guarded as adequate long term follow up series are lacking [1, 4].
References
1. Bosch EP, Mitsumoto H. Disorders of Peripheral Nerves. In: Neurology in Clinical Practice; The Neurological Disorders. Eds. Bradley WG, Daroff RB, Fenichel GM, Marsden CD. Vol-II, Second Edition, Newton, USA. Butterworth Heinemann Publication, 1996; pp 1881-1952.2. Donaghy M. Disorders of peripheral nerves. In: Brain's Diseases of the Nervous System. Ed. Walton J. Tenth Edition. New York, Oxford Medical Publications, 1993; pp 555-624.
3. Dyck PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dyck PJ, Thomas PK, Griffin JW, et al. (eds) Peripheral Neuropathy, 3rd ed. Philadelphia, W.B. Saunders, 1993, pp 1065-1093.
4. Donaghy M, Hakin RN, Bamford JM. Hereditary sensory neuropathy with neurotrophic keratitis. Description of an autosomal recessive disorder with a selective reduction of small unmyelinated nerve fibres and a discussion of the classification of the hereditary sensory neuropathies. Brain 1987; 110: 563-583.
5. Dyck PJ. Histologic measurements and fine structure of biopsied sural nerve: Normal and in peroneal muscular atrophy, hypertrophic neuropathy, and congenital sensory neuropathy. Mayo Clin Proc 1966; 41: 742-74.
6. Axelrod FB, Abularrage JJ. Familial dysautonomia: a prospective study of survival. J Pediatr. 1982 Aug;101(2):234-6. PubMed
7. Hilz MJ, Stemper B, Axelrod FB. Sympathetic skin response differentiates hereditary sensory autonomic neuropathies III and IV. Neurology 1999; 52: 1652-7. PubMed
8. Balachandran C, Sabitha L, Kantharaj GR. Hereditary sensory autonomic neuropathy - type-II in siblings. Indian J Lepr 1996; 373-374.
9. Jestico JV, Urray PA, Efphimiou J. A hereditary sensory and autonomic neuropathy transmitted as an X-linked recessive trait. J Neurol Neurosurg. Psychiat 1985; 48: 1259-1264. PubMed
10. Cavanagh NP, Eames RA, Galvin RJ, Brett EM, Kelly RE. Hereditary sensory neuropathy with spastic paraplegia. Brain 1979; 102: 79-94.
11. Kim JS, Woo YJ, Kim GM, et al. Congenital insensitivity to pain with anhidrosis: a case report. J Korean Med Sci 1999; 14: 460-4. PubMed
12. Sodaifi M, Aghaei S, Monabati A. Cutaneous variant of angiokeratoma corporis diffusum associated with angiokeratoma circumscriptum. Dermatol Online J. 2004 Jul 15;10(1):20. PubMed
13. Polo A, Aldegheri R, Bongiovanni LG, Cavallaro T, Rizzuto N. Painless fractures and thermoregulation disturbances in sensory-autonomic neuropathy; electrophysiological abnormalities and sural nerve biopsy. Neuropediatrics 2000; 31: 148-150. PubMed
14. Shah U, Arshad M, Mozaffar T. Dysphagia in hereditary sensory autonomic neuropathy type IV. J Pak Med Assoc 1999; 49: 121-123. PubMed
© 2006 Dermatology Online Journal