Kabuki syndrome: A new case associated with Becker nevus
Published Web Location
https://doi.org/10.5070/D37ff8819jMain Content
Kabuki syndrome: A new case associated with Becker nevus
Laura Cuesta MD, Isabel Betlloch MD, Fernando Toledo MD, Nuria Latorre MD, Almudena Flavia Monteagudo MD
Dermatology Online Journal 17 (8): 1
Department of Dermatology, Hospital General Universitario, Alicante, Spain. lcuestamontero@hotmail.comAbstract
Kabuki syndrome or Kabuki makeup syndrome was first described in 1981 in Japan by two different groups of authors. These investigators described a group of patients sharing typical facial features, skeletal anomalies, mental retardation, short stature, and dermatoglyphic anomalies. The term Kabuki makeup syndrome was coined because the peculiar facial features of the patients were reminiscent of the Japanese Kabuki theater masks. In 1988, Niikawa et al, after studying 62 patients, proposed five diagnostic criteria for this disease: peculiar facies (in 100% of all patients), skeletal anomalies (92%), dermatoglyphic anomalies (93%), medium to moderate mental retardation (92%), and short stature (83% of all cases). In addition to these findings, a variety of anomalies have been associated with this syndrome – the most serious being cardiac, renal, and urogenital abnormalities. We present a case of Kabuki syndrome in a 6-year-old boy who, in addition to the various features typical of the disease, also exhibited a Becker nevus - a condition not previously associated with this syndrome. The usefulness of dermoscopy in studying alterations in the dermatoglyphic patterns is also discussed.
Introduction
Kabuki syndrome or Kabuki makeup syndrome was first described in 1981 in Japan by Kuroki et al [1] and Niikawa et al [2]. These investigators described a group of patients sharing typical facial features, skeletal anomalies, mental retardation, short stature, and dermatoglyphic anomalies. The term Kabuki makeup syndrome was coined because the peculiar facial features of the patients were reminiscent of the Japanese Kabuki theater masks.
We present a case of Kabuki syndrome in a 6-year-old boy. In addition to the various features typical of the disease, he also exhibited Becker nevus - a condition not previously associated with Kabuki syndrome. The usefulness of dermoscopy in studying alterations in the dermatoglyphic patterns is also discussed.
Case report
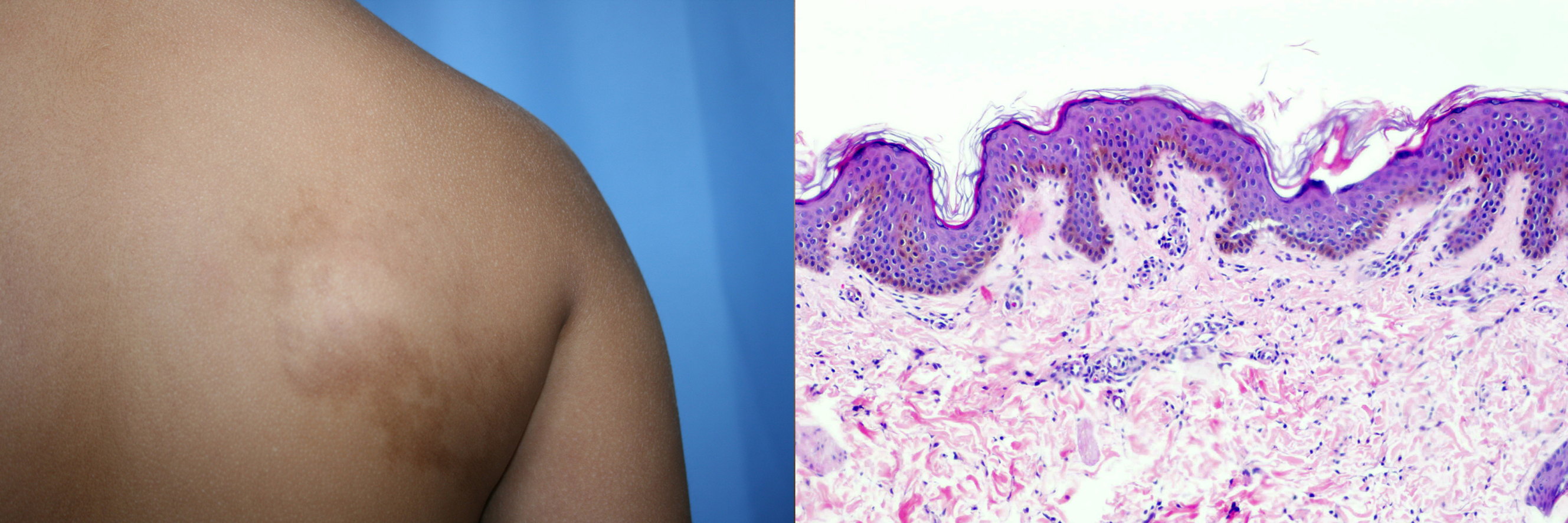 |
| Figure 1 |
|---|
| Figure 1. Hyperpigmented lesion containing hair on the right scapular region (at left). Biopsy from the lesion, showing acanthosis and elongation of the epidermal crests, hyperpigmentation of the basal layer and irregular and elongated smooth muscle fibers (at right). |
A 6-year-old boy, born of unrelated parents, was referred to our clinic because of a pigmented macule on the right buttock and another in the right scapular region. The mother was 32 years old and healthy. The child was born via a normal delivery in week 39. The postnatal period was normal. These hyperpigmented lesions had been present for years and had grown with the child; they were neither painful nor pruritic. Examination revealed two slightly hyperpigmented plaques containing scantly increased hair and exhibiting depressions in some zones (Figure 1). The clinical diagnosis was Becker nevus (Figure 1).
A biopsy showed acanthosis and elongation of the epidermal crests, with increased pigmentation of the basal layer and a large number of irregular, elongated smooth muscle fibers that confirmed the diagnosis of Becker nevus.
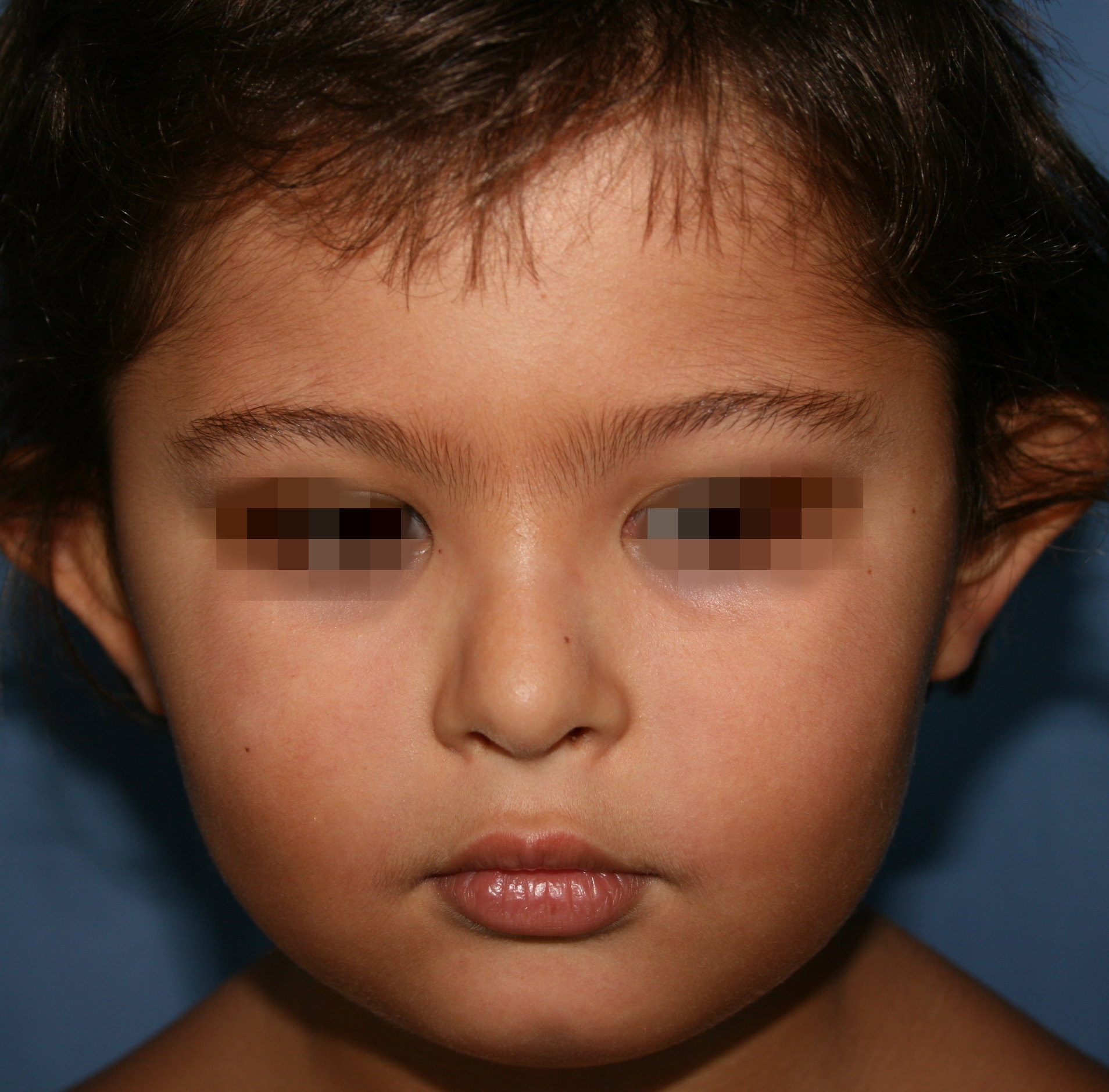 | 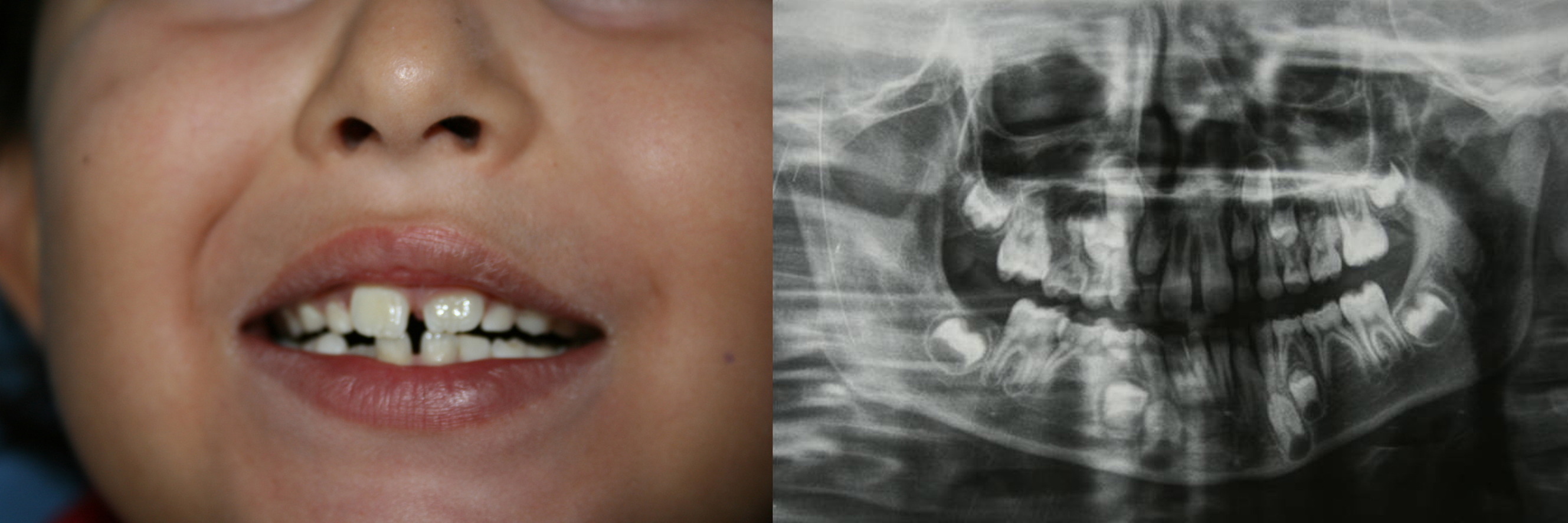 |
| Figure 2 | Figure 3 |
|---|---|
| Figure 2. The peculiar facies of the patient is shown. Note the arched eyebrows, the epicanthic fold, the elongated palpebral
fissures, and the dysmorphic and low-implanted ears. Figure 3. The patient had poorly aligned teeth (at left). The X-ray study showed agenesis of four premolars (at right). | |
However, the peculiar facial features of the patient were the most striking observation (Figure 2). Of note were the arched eyebrows, epicanthic fold, elongated palpebral fissures, a broad nasal base, dysmorphic and low-implanted ears, and poorly aligned teeth (Figure 3). The initial general examination also revealed a supernumerary nipple, scoliosis, a micropenis, excess body weight (> percentile 97) and a degree of mental retardation.
Given the characteristic facial features (very similar to Kabuki syndrome), we requested a series of complementary tests to confirm the syndrome. The pediatric neurologist confirmed marked hypotonus and the presence of a degree of mental retardation, which had been detected three years prior. The panoramic X-ray study showed agenesis of four premolars (Figure 3). Cryptorchidia and an elevator testicle were detected. The bone X-ray study showed shortening of the little finger of both hands secondary to shortening of the middle phalangeal bone (Figure 4). Genu varus and equinovarus were noted. The cardiological examination and electrocardiographic and echocardiographic findings were normal. The karyotype corresponded to 46XY. The endocrine study likewise proved normal.
 |  |
| Figure 4 | Figure 5 |
|---|---|
| Figure 4. The patient had shortening of the little finger of the hands clinically (at left); the bone X-ray study showed shortening
of the little finger of both hands secondary to shortening of the middle phalangeal bone (at right). Figure 5. The patient showed an increased ulnar loop and fingertip pads (at left). We used dermoscopy to improve the diagnosis of the dermatoglyphic alterations (at right). | |
Fingertip pads were noted on several fingers, as well as an increased digital ulnar loop pattern. Indian ink and dermoscopy were used to assess the dermatoglyphic alterations (Figure 5). Of note is the fact that we used dermoscopy to improve the diagnosis of these dermatoglyphic alterations, which may be difficult to recognize by simple inspection.
Discussion
Kabuki syndrome (KS) is a rare disorder described in 1982 [1, 2]. Five diagnostic criteria have been proposed, such as characteristic facial features (in 100% of all patients), skeletal anomalies (92%), dermatoglyphic alterations (93%), mental retardation (92%), and short stature (83%). The syndrome is more common in Japan, where the estimated prevalence is 1/32,000 live births [3, 4]. To date, about 400 cases have been reported worldwide [4] and fewer than 30 cases in Spain since 1995 [5, 6, 7] .
The most important diagnostic criterion is the characteristic patient facies [4], with arched eyebrows, epicanthic fold, lower palpebral eversion, elongated palpebral fissure, ptosis, strabismus, a short nasal septum, prominent and malformed ears, abnormal dentition, an arched or cleft palate, micrognathia, pits in the lower lip, and a low hair implantation line. The dental alterations have been recently reviewed and noted to be an important feature in addition to the five main characteristics of KS [4]. Our patient showed most of the facial alterations, although the frequently described palatal abnormalities were not observed [4].
The most frequent skeletal anomalies are shortening of the little finger and clinodactyly, together with scoliosis, vertebral anomalies, hip dislocation, foot deformities, and even occult spina bifida [4, 8]. Our patient presented with brachydactyly of the little finger secondary to shortening of the middle phalangeal bone, scoliosis, equinovarus, and genu varus.
The dermatoglyphic alterations described in 1982 [9] include increased ulnar loops (the most common finding), absence of digital triradius c or d, presence of interdigital triradius bc or cd, hypothenar loop pattern, and ulnar loop pattern in the fourth interdigital area [3, 4, 9]. In addition, the presence of fingertip pads has been reported. For studying the dermatoglyphic alterations, we used dermoscopy with Indian ink. Our patient showed increased ulnar loops as well as the presence of fetal pads on several fingertips.
Mental retardation is the only neurological anomaly described as a diagnostic criterion. However, one-sixth of patients have a normal intelligence quotient [4]. Hypotonus has also been observed and included in the differential diagnosis of neonatal hypotonus [10]. Our patient exhibited slight psychomotor retardation from infancy, although his adjustment to school was normal. Significant hypotonus was present, limiting physical activity.
The last diagnostic criterion is short stature [3, 4], with growth retardation manifesting after three months [4]. Obesity is seen in almost 20 percent of all cases [4]. This necessitates a differential diagnosis that includes other syndromes that combine obesity with mental retardation in infancy, such as Prader Willi syndrome [11]. Growth hormone anomalies [12] and early puberty have also been documented. Our patient presented with normal percentiles in length, but body weight was above the 97th percentile without hormone alterations.
Internal organ anomalies have been described. The most frequent cardiac anomaly is aortic coarctation followed by septal alterations [4, 13]. Recurrent pneumonia appears to be the most common respiratory problem [4]. Renal and urogenital alterations are seen in 25 to 28 percent of all patients [4, 8]. They include renal malformations – ureteral alterations, cryptorchidia (24%), and micropenis (10%). Gastrointestinal alterations appear in less than 5 percent of patients [4]. In our case we observed cryptorchidia with elevated testicle and a micropenis. The renal-urological study and cardiological examinations proved normal and there were no pulmonary or digestive tract manifestations.
An increase in infections (63%), immune deficiencies, and hematological disorders have been described [3, 4]. In our patient, however, the immune and autoimmune tests proved normal.
There have been descriptions of a number of ectodermal alterations, including fragile nails, trichorrhexis nodosa, irregular hair diameter, and congenital alopecia areata [14, 15], not seen in our patient. However, a supernumerary nipple was noted (also previously reported) [7]. Hyperpigmented lesions associated with KS have been described in one patient [14]. Our review of the literature yielded no previous reports of the association between Becker nevus and KS as in our patient. Ours is the first case documenting hyperpigmentation with a histological evaluation.
The diagnosis of KS is based on clinical criteria, because no genetic alterations have been established. Most cases are sporadic [4, 8], although some of them present in a familial grouping. Multiple chromosomal alterations have been described, but none of them serve to define Kabuki syndrome [4]. The karyotype was normal in our patient and none of the relatives presented similar clinical characteristics.
Conclusion
Kabuki syndrome is a rare multiple malformation syndrome, probably more common than assumed. Kabuki syndrome is more prevalent in Japan and few Spanish cases have been documented. Dermatologists have an important role alongside pediatricians in identifying Kabuki syndrome.
References
1. Kuroki Y, Suzuki Y, Chyo H et al. A new malformation síndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal and mental retardation. J Pediatr 1981;99:570-73. [PubMed]2. Niikawa N, Matsuura N. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 1981; 99:565-69. [PubMed]
3. Niikawa N, Kuroki Y, Kajii T et al. Kabuki make-up (Niikawa-Kuroki) síndrome: a study of 62 patients. Am J Med Genet. 1988; 31:565-589. [PubMed]
4. Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet 2003;117:57-65. [PubMed]
5. Galán-Gómez E, Cardesa-García JJ, Campo-Sampedro F et al. Kabuki make-up (Niikawaa-Kuroki) syndrome in five spanish children. Am J Med Genet 1995; 59:276-282. [PubMed]
6. González Armengod C, García-Alix A, del Campo M et al. Kabuki's syndrome: a recognizable picture from early infancy. An Esp Pediatr 1997;47:429-431. [PubMed]
7. Pascual-Castroviejo I, Pascual-Pascual SI, Velázquez-Fragua et al. Kabuki make-up síndrome. A report of 18 Spanish cases. Rev Neurol 2005;40:473-478. [PubMed]
8. Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet 2005;67:209-219. [PubMed]
9. Niikawa N, Kuroki Y, Kajh T. The dermatoglyphic pattern of the Kabuki make-up syndrome. Clinical Genetics 1982;21:315-320. [PubMed]
10. Aguilera S, Botella MP, Ocio I. Kabuki syndrome in the differential diagnosis of neonatal hypotonia. An Pediatr (Barc.) 2009;70:91-93.
11. Battin J. Genetic obesity in childhood. Bull Acad Natl Med 2009; 193:1281-8. [PubMed]
12. Tawa R, kaino Y, Ito I et al. A case of kabuki make-up síndrome with central diabetes insipidus and growth hormone neurosecretory dysfunction. Acta Paeditr Jpn 1994; 36:412-415. [PubMed]
13. Digilio MC, Marino B, Toscano A et al. Congenital heart defects in kabuki syndrome. Am J Med Genet 2001; 100;269-274. [PubMed]
14. Bereket A, Turan S, Alper G et al. Two patients with Kabuki syndrome presenting with endocrine problems. J Pediatr Endocrinol Metab 2001;14:215-20. [PubMed]
15. Lerone M, Priolo M, Naselli A et al. Ectodermal abnormalities in Kabuki syndrome. Am J Med Genet 1997;73:263-266. [PubMed]
© 2011 Dermatology Online Journal