Dyschromatosis universalis hereditaria: A rare entity
Published Web Location
https://doi.org/10.5070/D37dr7d2h2Main Content
Dyschromatosis universalis hereditaria: A rare entity
Sumir Kumar MD, Bharat Bhushan Mahajan MD, Rajwinder Singh Jr
Dermatology Online Journal 17 (7): 6
Department of Dermatology, Venereology and Leprology, Guru Gobind Singh Medical College and Hospital, Faridkot, PunjabAbstract
Dyschromatosis universalis hereditaria is an infrequently occurring genodermatosis with peculiar pigmentary changes, consisting of varying sized, intermingled hyperpigmented and hypopigmented macules that give an overall impression of mottling. We hereby report a case of dyschromatosis universalis hereditaria in a young female with a family history of the disorder.
Introduction
Dyschromatoses are a group of disorders characterized by the presence of both hyperpigmented and hypopigmented macules, many of which are small in size and irregular in shape. These are a spectrum of diseases, which includes dyschromatosis universalis hereditaria (DUH), dyschromatosis symmetrica hereditaria (DSH), acropigmentation of Dohi, Dowling-Degos disease, and a segmental form called unilateral dermatomal pigmentary dermatosis (UDPD). Dyschromatosis symmetrica hereditaria (DSH) was first reported as a clinical entity by Toyama in 1929 [1]. It is characterized by a symmetrical distribution of hyperpigmented and hypopigmented macules on the extremities, especially over the dorsa of the hands and feet. In 1933 Ichikawa and Hiraga described DUH – which was essentially the same disorder but occurring in a generalized distribution – presenting as a generalized leukomelanoderma with relative sparing of the face, palms, and soles [2]. We hereby report a case of dyschromatosis universalis hereditaria in a young Indian female with family history of the disorder.
Case report
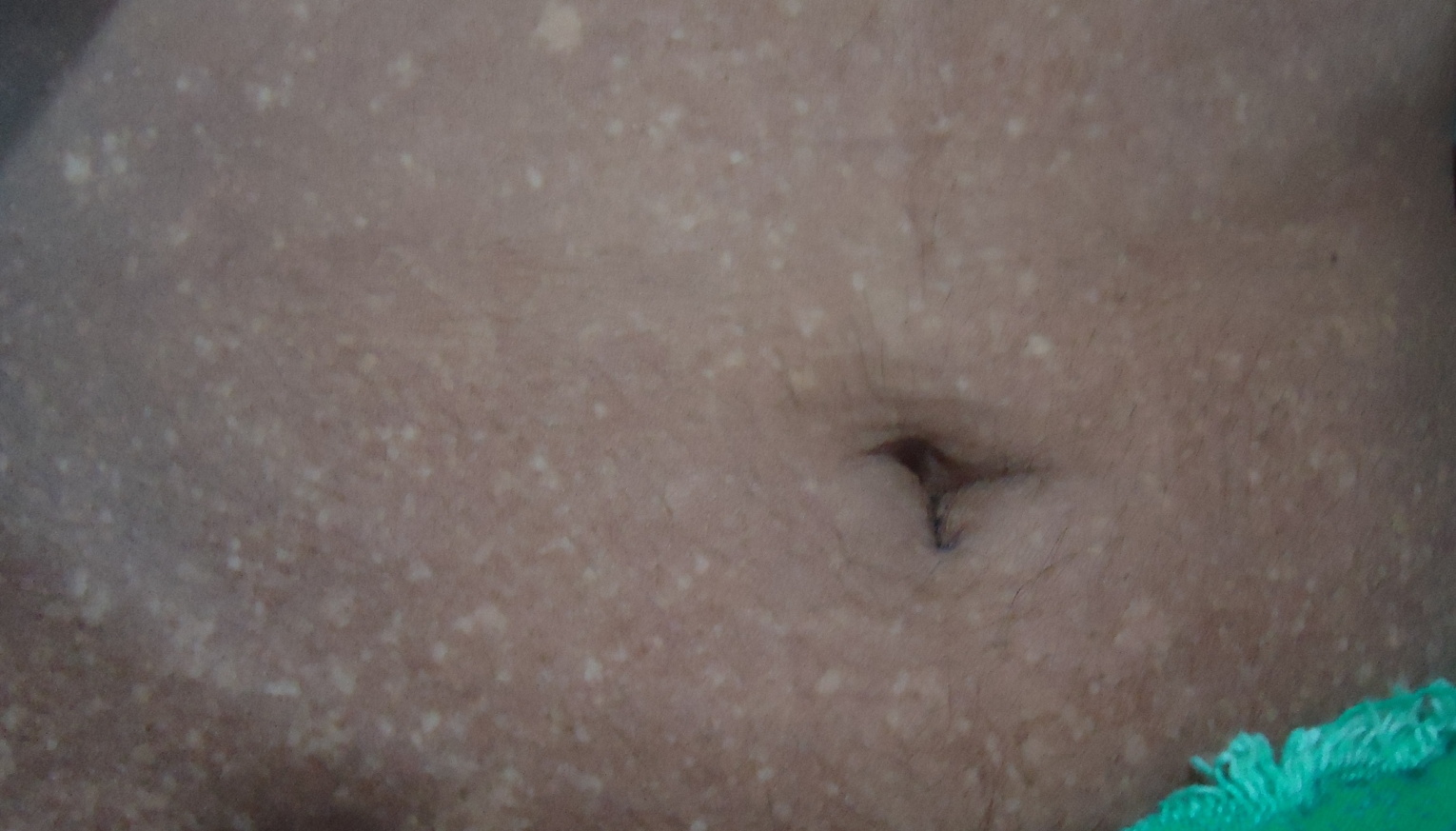 |  |
| Figure 1 | Figure 2 |
|---|
A 22-year-old unmarried female presented to us with a chief complaint of multiple hyperpigmented and hypopigmented macules over arms, legs, and trunk for the past 15 years. The lesions had started over both the forearms and legs and gradually spread upwards towards the thighs, arms, and then the trunk over a period of 5 years. There was no history of photosensitivity. There was no history of handling any chemical directly or of any significant history of drug intake. There was a history of similar lesions in an elder brother. He, however, could not be examined because he migrated abroad 2 years previously. There was no history of consanguinity among the parents.
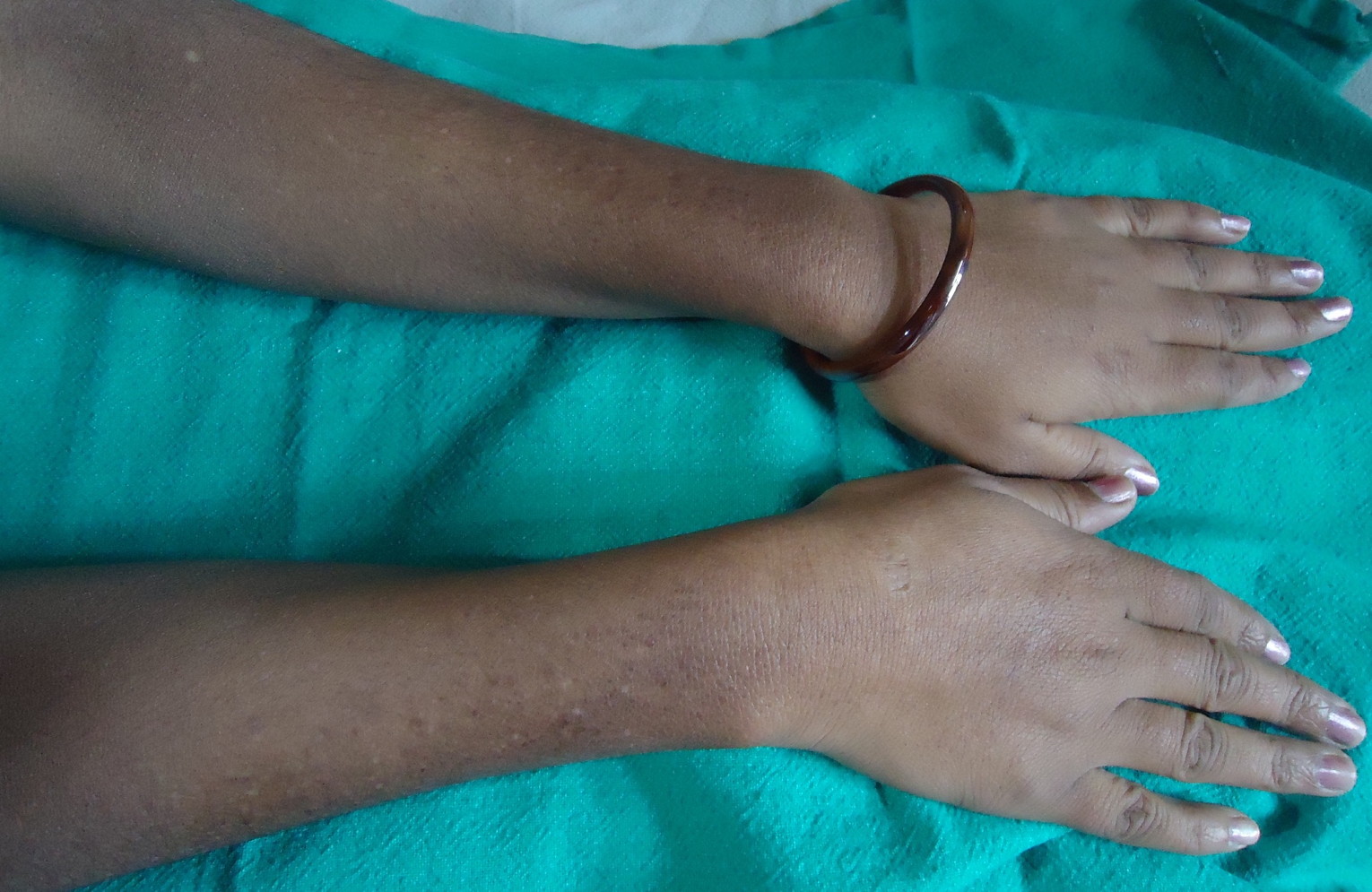 |  |
| Figure 3 | Figure 4 |
|---|
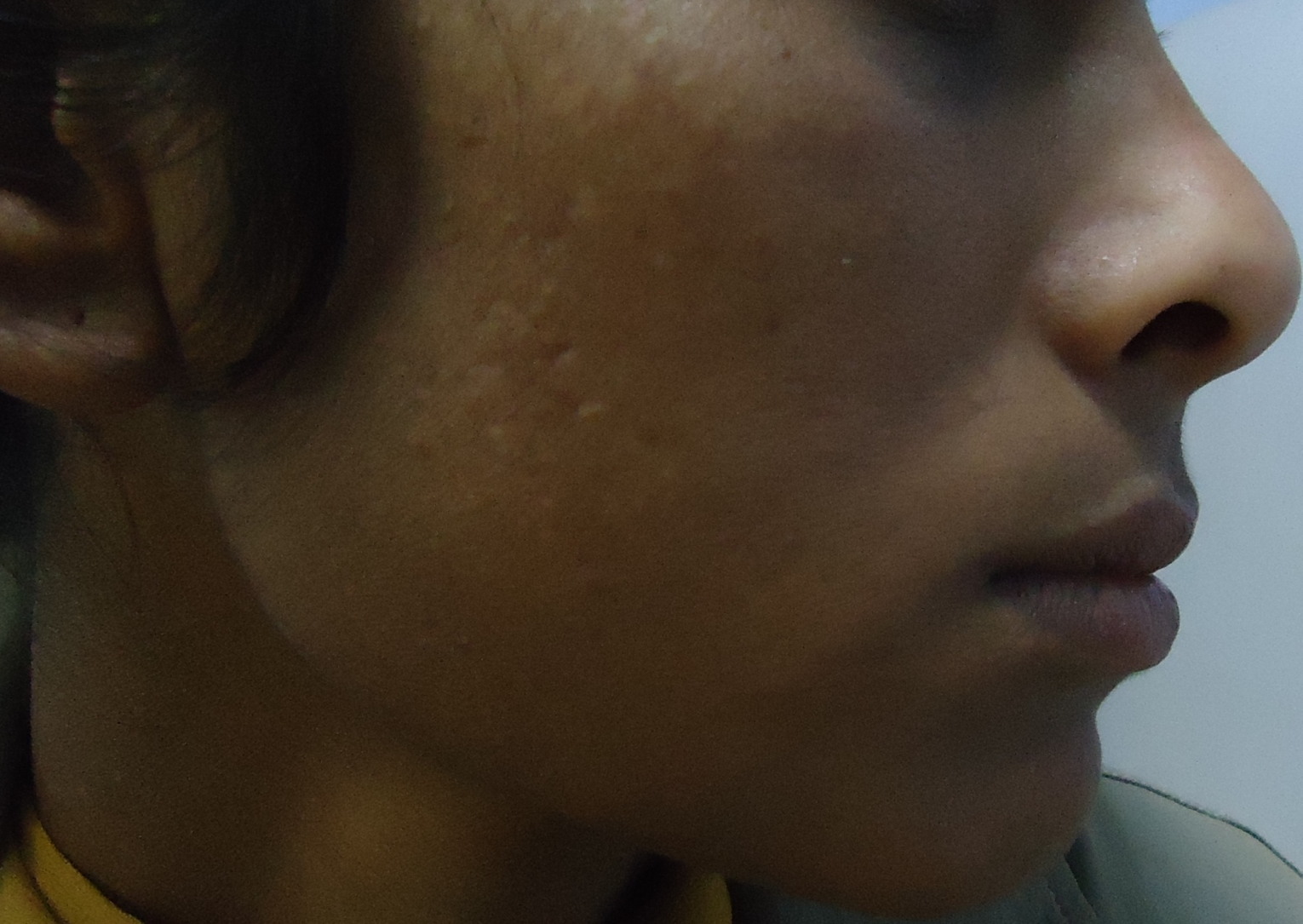 |
| Figure 5 |
|---|
On dermatological examination multiple hyperpigmented and hypopigmented macules were present in a reticulate pattern on the trunk (Figures 1 and 2), arms, forearms (Figure 3), thighs, and legs (Figure 4). A few hypopigmented macular lesions were also present on the face (Figure 5). Her palms, soles, and mucous membranes were within normal limits. Systemic examination did not reveal any abnormality. Routine laboratory investigations, including blood count, urine C/E, liver function tests, renal function tests, and electrolytes, were within normal limits. VDRL and ELISA for HIV were non reactive.
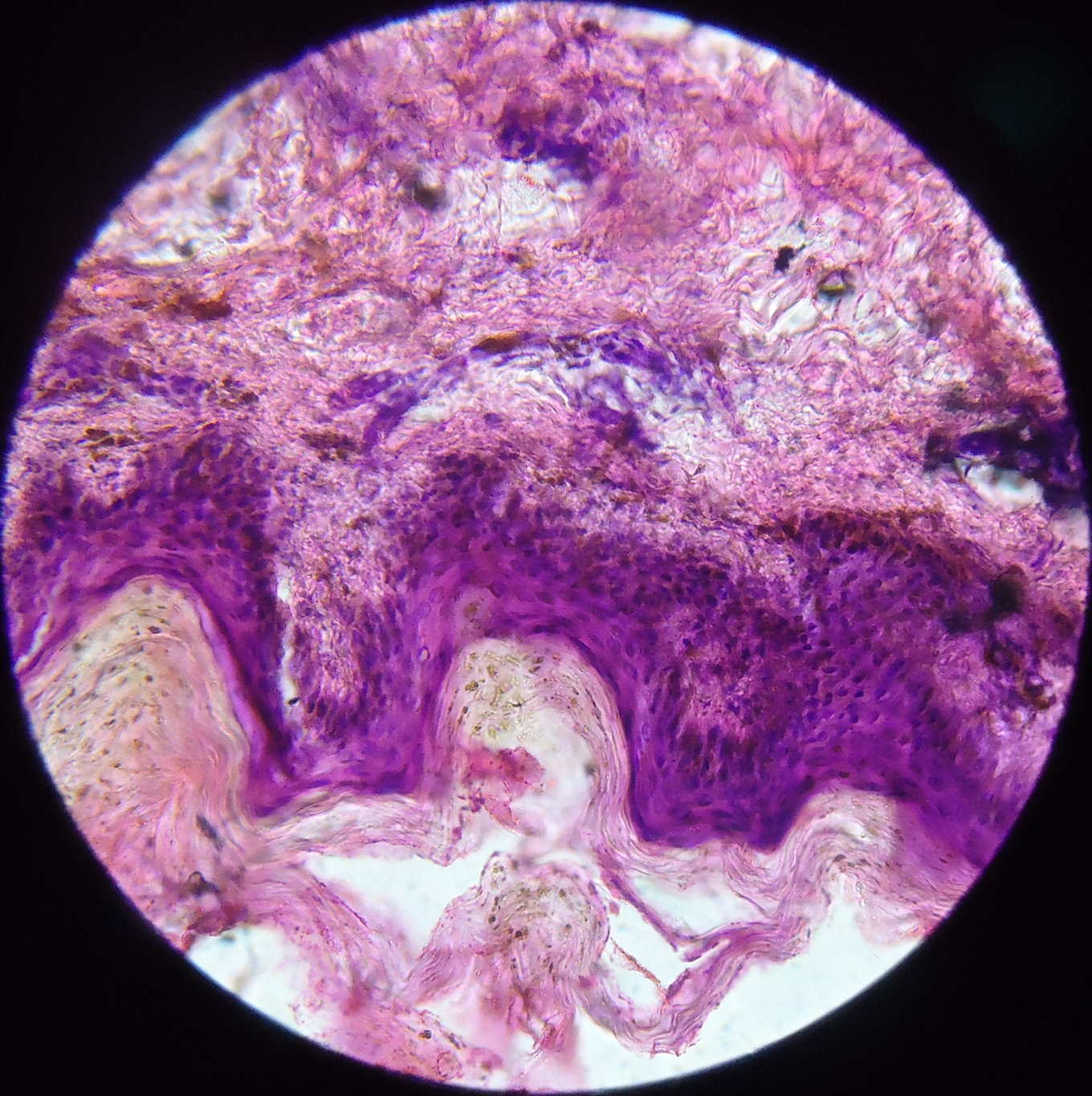 | 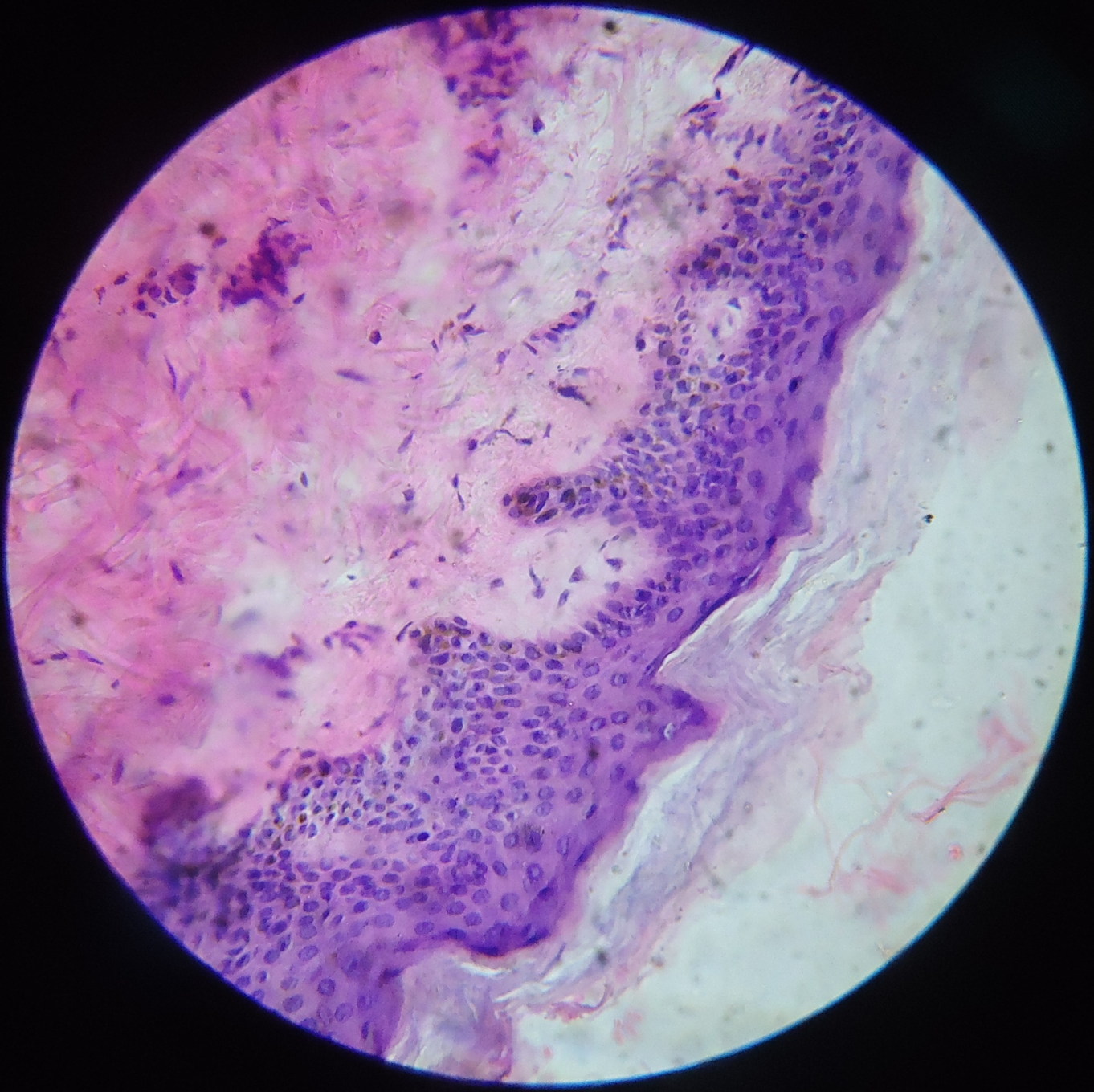 |
| Figure 6 | Figure 7 |
|---|
A skin biopsy was taken from both the hyperpigmented and hypopigmented lesions. The basal layer in the hyperpigmented lesions showed an increase in pigment along with melanin incontinence (Figure 6) whereas the hypopigmented lesions it showed a decrease in pigment (Figure 7). The dermis in both the biopsies showed mild perivascular lympho-mononuclear infiltrate.
Discussion
Dyschromatosis universalis heriditaria is a rare genodermatosis that is commonly encountered in Japan. However, rare familial cases have been reported from Europe [3], China [4] and India [5, 6]. Dyschromatosis universalis heriditaria is an autosomal dominant disorder with variable penetrance, but a few individuals have inherited it in an autosomal recessive fashion [2, 7]. Although the precise etiology of this disorder is not yet known, the DUH locus has recently been mapped to chromosome 6q24.2-q25.2 [8]. In the case of our patient there was a history of similar lesions in an elder brother.
It has been suggested in the past that DUH is a disorder of melanocyte number. However, based on a recent electron microscopic study it has been suggested that DUH may be a disorder of melanosome production in epidermal melanin units rather than a disorder of melanocyte number [9].
Dyschromatosis universalis hereditaria may be associated with abnormalities of dermal connective tissue or nerve tissue; it may be associated with other systemic complications [7, 10]. No such features characterized our patients. Abnormalities of hair and nails have also been reported, but such abnormalities were not present in our patient [11].
Lesions of dyschromatosis symmetrica hereditaria (DSH) have to be differentiated from xeroderma pigmentosum because in both the disorders patients clinically show lesions in the photoexposed areas. However, in our patient, lesions were present in the unexposed sites as well. Moreover, the lesions did not show any atrophy or telangiectasia as seen in xeroderma pigmentosum. Other diseases in the differential diagnosis include dyschromic amyloidosis and exposure to chemicals such as diphenylcyclopropenone and monobenzyl ether of hydroquinone [1, 9]. Yet, no such history of contact was present in our patient. Pinta in late stages can present similarly, but there was no history of any lesion suggestive of primary and secondary (pintids) lesions of pinta. Vitiligo with areas of repigmentation can mimic this condition, but generalized involvement and histopathology ruled out vitiligo.
No treatment modality is available. Only genetic counseling is advised because of the recent reports of its genetic etiology. Generally, DUH does not progress or worsen with age. In our case there was no spontaneous regression, and, although there was a slight progression in the initial stages, the disease has been stable for past 10 years.
Conclusion
This case of DUH is reported because of its rarity. Despite its rarity it assumes significance as it must be distinguished from xeroderma pigmentosum and other dyschromias.
References
1. Toyama J. Dyschromatosis symmetrica hereditaria. Jap J Dermatol 1929; 29: 95-96.2. Urabe K, Hori Y. Dyschromatosis. Semin Cutan Med Surg 1997;16:81-5. [PubMed]
3. Rycroft RJ, Calnan CD, Wells RS. Universal dyschromatosis with small stature and high tone deafness. Clin Exp Dermatol 1977;2:45-8. [PubMed]
4. Wang G, Li CY, Gao TW, Liu YF. Dyschromatosis universalis hereditaria: Two cases in a Chinese family. Clin Exp Dermatol 2005;30:494-6. [PubMed]
5. Gharpuray MB, Tolat SN, Patwardham SP. Dyschromatosis: Its occurrence in two Indian families with unusual features. Int J Dermatol 1994;33:391-2. [PubMed]
6. Kantharaj GR, Siddalingappa K, Chidambara Murthy S. Dyschromatosis universalis: Autosomal dominant pattern. Indian J Dermatol Venereol Leprol 2002;68:50-1. [PubMed]
7. Bukhari IA, EL-Harith EA, Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. J Eur Acad Dermatol Venereol 2006;20: 628-629. [PubMed]
8. Xing Q, Wang M, Chen X, et al. A gene locus responsible for dyschromatosis symmetrica hereditaria (DSH) maps to chromosome 6q24.2-q25.2. Am J Hum Genet 2003;73: 377-382. [PubMed]
9. Kim NS, Im S, Kim SC. Dyschromatosis universalis hereditaria. J Dermatol 1997;24:161-164. [PubMed]
10. Al Hawsawi K, Al Aboud K, Ramesh V, Al Aboud D. Dyschromatosis universalis hereditaria: report of a case and review of the literature. Pediatr Dermatol. 2002;19:523-526. [PubMed]
11. Sethuraman G, D'Souza M, Mohan Thappa D, Srinivas CR, Smilest L. Clin Exp Dermatol 2002;27:477-479. [PubMed]
© 2011 Dermatology Online Journal