Hypohidrotic ectodermal dysplasia
Published Web Location
https://doi.org/10.5070/D37021x2grMain Content
Hypohidrotic ectodermal dysplasia
Phoebe D Lu MD PhD, Julie V Schaffer MD
Dermatology Online Journal 14 (10): 22
Department of Dermatology, New York UniversityAbstract
We report three children with hypohidrotic ectodermal dysplasia (HED), which includes two sisters with unaffected parents (and therefore likely autosomal recessive inheritance of HED) and an unrelated boy. Each patient presented with hypohidrosis, sparse hair, oligodontia with conical teeth, periorbital hyperpigmentation, eczematous dermatitis, and facial features that include frontal bossing, a saddle nose, and prominent lips. HED is caused by defects in the ectodysplasin signal transduction pathway. Mutations in the gene encoding the ligand ectodysplasin A (EDA) underlie classic, X-linked recessive HED, whereas mutations in the genes encoding the EDA receptor and (less frequently) the adaptor protein that associates with the EDA receptor's death domain result in autosomal dominant and autosomal recessive forms of HED.
 |  |
| Figure 1 | Figure 2 |
|---|
History
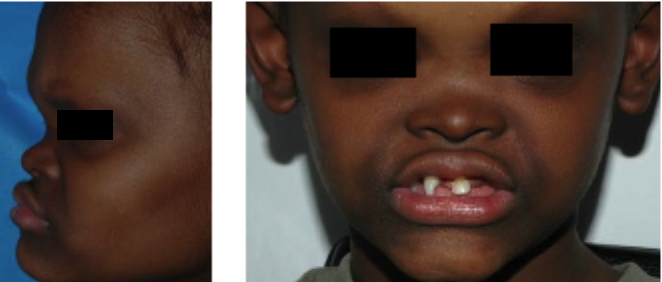
A.A. presented to the Pediatric Dermatology Clinic at Bellevue Hospital Center in September, 2001, with eczematous dermatitis on the forearms. He had a history of recurrent high fevers during infancy, and his adoptive mother reported that he does not sweat. To prevent overheating, he has worn a cooling vest while participating in outdoor activities during warm weather. Medical history includes oligodontia, chronic nasal congestion, impacted cerumen, and mild conductive hearing loss. Medical interventions have included topical glucocorticoid therapy and the use of a dental prosthesis. He had no family history of hypohidrosis, hypotrichosis, or dental abnormalities; his fraternal twin brother is unaffected.
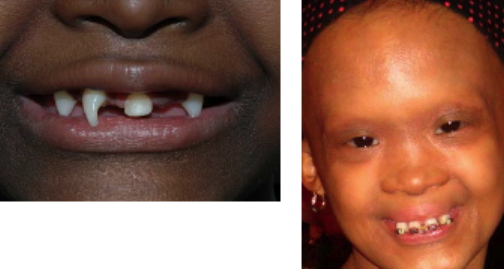
T.V. and C.V. presented to the Pediatric Dermatology Clinic at the Charles C. Harris Skin and Cancer Pavilion in July, 2005, with eczematous dermatitis on the extremities. T.V. had a history of recurrent high fevers during infancy, and the patients' mother reported that neither of the girls is able to sweat and required precautions to avoid overheating during warm weather and physical exertion. Both sisters had oligodontia, which has been treated with prostheses and dental reconstruction. In addition, T.V. had surgical correction of strabismus, and C.V. had surgical correction of cryptotia. Their dermatitis has been managed with intermittent topical glucocorticoid therapy. No other family members (including T.V.'s fraternal twin sister) were affected by hypohidrosis, hypotrichosis, or dental abnormalities.
Physical Examination
All three children had fine, sparse scalp hair, eyebrows, and eyelashes. Their skin was smooth and dry, and periorbital (and, in A.A., perioral) hyperpigmentation and wrinkling were evident. Sebaceous hyperplasia was apparent in various degrees on the medial aspects of the cheeks, nose, and chin. The teeth were reduced in number and conical in shape, the alveolar ridges were underdeveloped, and the lips were prominent. Additional facial features shared by the three children included frontal bossing, prominent supraorbital ridges, and midfacial hypoplasia with a depressed nasal root and bridge.
Lab
None.
Histopathology
None.
Comment
Ectodermal dysplasias are genetic disorders that result in defective structure or function of two or more major derivatives of the ectoderm, which include the sweat glands, hair, teeth, and nails. Hypohidrotic ectodermal dysplasia (HED; Christ-Siemens-Touraine syndrome) represents a group of ectodermal dysplasias that are characterized by sparse or absent eccrine glands as well as by hypotrichosis and oligodontia with peg-shaped teeth [1]. Because of their severely diminished ability to sweat, patients with HED have a propensity to develop hyperthermia with physical exertion or exposure to a warm environment, and affected infants often present with recurrent high fevers. The scalp hair, eyebrows, and eyelashes are sparse, fine, and oftentimes lightly pigmented. In contrast to several other types of ectodermal dysplasia, the nails are normal. Additional cutaneous features of HED include scaling or peeling of the skin during the neonatal period, periorbital hyperpigmentation and wrinkles, facial sebaceous hyperplasia, and eczematous dermatitis. Hypohidrotic ectodermal dysplasia patients have a characteristic facies with frontal bossing, a saddle nose, and full, everted lips. Abnormal mucous glands result in extremely thick nasal secretions and a propensity to develop respiratory tract infections.
 |
| Figure 3 |
|---|
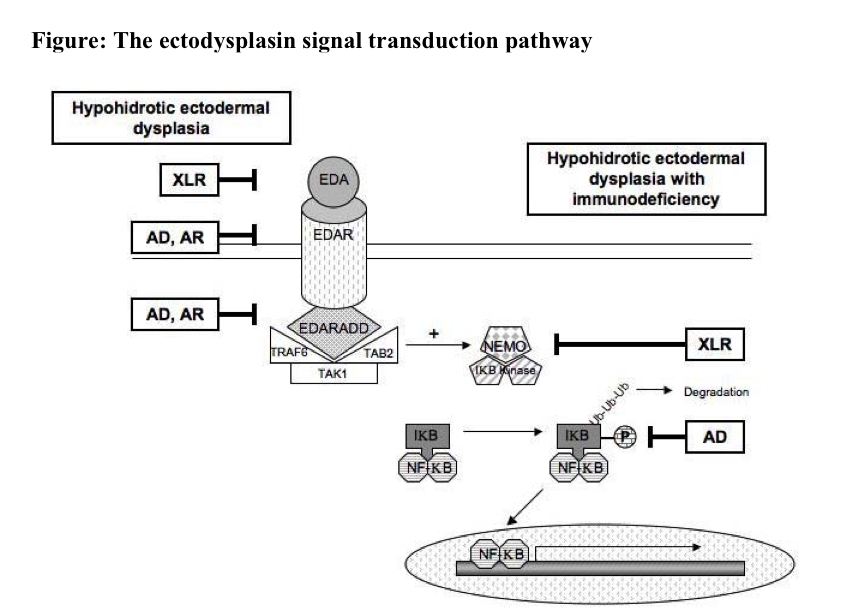
Hypohidrotic ectodermal dysplasia is caused by mutations in genes that encode several proteins with roles in the ectodysplasin signal transduction pathway (Figure 3). Activation of this cascade within epithelial cells at a critical time during embryogenesis results in translocation of the transcription factor NF-κB into the nucleus and subsequent expression of target genes that are involved in the morphogenesis of eccrine sweat glands, hair follicles, and teeth. Mutations in the EDA gene, which encodes the ectodysplasin ligand that initiates signaling through this pathway, cause the X-linked recessive (XLR) form of HED that accounts for 75 to 95 percent of HED cases [2]. Ectodysplasin is a transmembrane protein belonging to the tumor necrosis factor (TNF) family of ligands and exists in two isoforms, ectodysplasin A1 and A2. Cleavage of ectodysplasin A1 by furin generates soluble ligand that binds to its receptor, EDAR, on epithelial cells. Mutations in EDA affect the processing and trimerization of ectodysplasin A1 and its ability to interact with EDAR.
Mutations in the EDAR gene can be identified in approximately 25 percent of HED patients, who do not have an EDA defect [3]. EDAR is a transmembrane protein in the TNF receptor family, and its intracellular portion contains a death domain that interacts with an adaptor protein, EDARADD. Dominant-negative EDAR mutations (e.g., those that affect receptor trimerization) produce an autosomal dominant (AD) form of HED, while other EDAR mutations (e.g., those that lead to a premature termination codon and subsequent nonsense-mediated mRNA decay) result in an autosomal recessive (AR) form of HED. Mutations in the EDARADD gene also may underlie both AD and AR forms of HED [4]. In addition, HED associated with immunodeficiency may occur in XLR and AD forms, which are caused by mutations in the NF-κB essential modulator (NEMO) gene and inhibitor of κB-α (IKBA) gene, respectively. The NEMO mutations are hypomorphic, which impair but do not abolish NEMO protein function, whereas the IKBA mutations are hypermorphic and lead to increased IKBA function by preventing the phosphorylation and subsequent ubiquitination and degradation of this protein; however, both defects result in decreased NF-κB signaling.
Management of HED includes external cooling and regulation of ambient temperatures to avoid hyperthermia, dental restoration, and treatment of associated eczematous dermatitis and respiratory tract infections. In mice and dogs with XLR ectodermal dysplasia due to Eda mutations, administration of recombinant ectodysplasin A1 immediately after birth or (in mice) in utero can partially or completely correct the phenotype, which suggests a potential future therapy for HED [5].
References
1. Cui C-Y, Schlessinger D. EDA signaling and skin appendage development. Cell Cycle 2006; 5: 2477 PubMed2. Kere J, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13: 1996; 409 PubMed
3. Van der Hout AH, et al. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur J Human Genetics 2008; advance online publication, doi:10.1038/sj.ejhg.5202012 PubMed
4. Bal E, et al. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum Mutat 2007; 28: 703 PubMed
5. Gaide O, Schneider P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med 2003; 9: 614 PubMed
© 2008 Dermatology Online Journal