Hyperimmunoglobulin E syndrome
Published Web Location
https://doi.org/10.5070/D36q44b3tbMain Content
Hyperimmunoglobulin E syndrome
Benjamin E Rosenberg MD
Dermatology Online Journal 10 (3): 4
From the Ronald O. Perelman Department of Dermatology, New York University
Abstract
A 63-year-old man presented with erythroderma, peripheral blood eosinophilia, elevated serum IgE levels, and a history of dermatitis, furunculosis, and a cold abscess. Hyperimmunoglobulin E syndrome is a rare, multisystem disorder that is characterized by cutaneous and sinopulmonary infections, dermatitis, and elevated serum IgE levels. Traditional therapy includes good skin care, antibiotics for skin and pulmonary infections, incision and drainage of abscesses, and emollients plus topical glucocorticoids for dermatitis.
Clinical synopsis
History.—A 63-year-old man presented with a dermatosis that began in childhood. His skin problems began as multiple, exudative groin eruptions that often became superinfected. During his teenage years, these eruptions continued and he also experienced episodes of furunculosis involving the buttocks, groin, and thighs. At that time it was not uncommon for him to have over 20 furuncles at once. He also noted a propensity to develop cutaneous fungal infections of the groin and elsewhere on his skin. He developed a cold abscess on his left elbow in 1996, which was treated with incision and drainage as well as with intravenous and oral antibiotics. In February 2003 the patient presented to the New York Harborview Health Care Dermatology Clinic with erythroderma, which evolved into a subacute eczematous dermatitis involving the trunk and proximal extremities after treatment with clobetasol propionate (0.1% ointment), cephalexin, emollients, and antihistamines. Although he denies a history of recurrent pneumonias, he has experienced chronic bronchitis. He has no dental or joint problems but does have a history of fractures of the left arm and left foot. He denies a history of asthma or allergic rhinitis. Past medical history includes a diffuse, poorly differentiated large-cell lymphoma, which was treated with chemotherapy in 1976 with remission and no evidence of recurrence, and hypertension that was treated with lisinopril. He takes aspirin daily. Family history includes furunculosis in the patient's mother, but there is no history of asthma, allergic rhinitis, or atopic dermatitis.
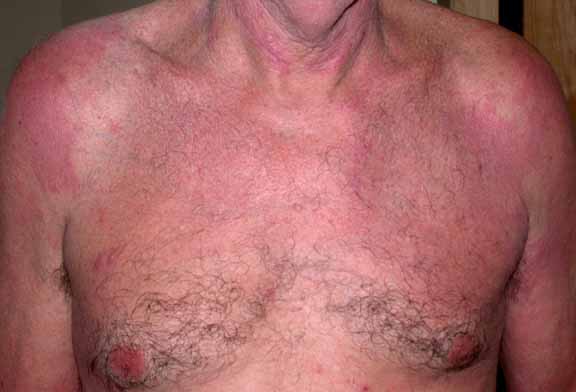
Physical Examination.—Discrete, erythematous, excoriated papules were present on the posterior and left-lateral aspect of the neck. Lichenified plaques were noted on the trunk and the posterior aspects of the arms. On the buttocks were multiple, firm, lichenified nodules, and there were erythematous papules on the thighs. The facial features, joints, and teeth were normal.
|
|
| Figure 1 | Figure 2 |
|---|
Laboratory Data.—A complete blood count showed 10 percent eosinophils on the differential analysis, erythrocyte sedimentation rate was elevated at 39 mm/hr, and immunoglobulin E levels were greater than 100,000 U/L. Immunoglobulins G, A, and M, the basic metabolic profile, and hepatic function tests were normal.
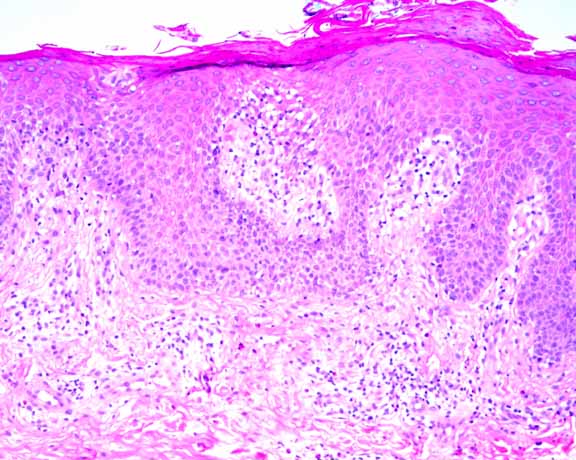
Histopathology.—There is a perivascular infiltrate of lymphocytes, macrophages, and eosinophils. Lymphocytes extend to the epidermis where there is psoriasiform epidermal hyperplasia, spongiosis, and overlying parakeratosis. The periodic-acid-Schiff stain is negative for fungi.
Diagnosis.—Hyperimmunoglobulin E syndrome.
Comment
The hyperimmunoglobulin E syndrome (HIE) is a rare, idiopathic, multisystem disorder with immunodeficiency [1, 2]. It was first described by Davis et al. in 1966 in two girls with red hair, chronic dermatitis, and recurrent staphylococcal abscesses and pneumonias. Inspired by the biblical character whose body was smitten with boils by Satan, they referred to this condition as Job's syndrome [2, 3]. In 1972, Buckley et al. described two boys with similar symptoms as well as a coarse facies, peripheral blood eosinophilia, and elevated serum IgE levels [3]. Since that time, over 200 hundred cases have been reported in the literature [1].
The classic triad of HIE includes recurrent cutaneous and sinopulmonary infections, chronic dermatitis from birth or early childhood, and elevated serum IgE levels [1, 2, 3, 4]. The cutaneous infections, which are predominantly caused by Staphyloccus aureus, include pustules, furuncles, abscesses, cellulitis, lymphangitis, and paronychias. Cold abscesses, which are fluctuant, nontender, nonerythematous masses caused by S. aureus, are pathognomonic for HIE; however, they are not required for the diagnosis. Candidal and dermatophyte infections of the skin, nails, and mucosae are also common [1, 2, 3, 4]. The sinopulmonary infections are also usually caused by S. aureus infection, but may be caused by Hemophilus influenzae or other bacteria; these infections can lead to empyemas, bronchiectasis, or pneumatoceles [1, 2, 3]. The pruritic dermatitis in HIE often develops in infancy or early childhood, and may be confused with atopic dermatitis. However, the patients lack a history of asthma, allergic rhinitis, or a family history of atopy [1, 2, 3]. Biopsy specimens show an eosinophilic infiltrate [2]. Serum IgE levels in HIE are elevated over ten times the normal limit or greater than 2000 IU/mL, but there is no correlation between the IgE levels and the severity of the disease. The IgE levels tend to remain constant during the course of the disease [2, 4]. The patients often demonstrate a peripheral eosinophilia of their blood and sputum [2]. Other features include a characteristic facies with frontal bossing, a broad nasal bridge, a wide, fleshy nasal tip, and deep-set eyes [2, 3, 4]. Bony abnormalities include osteopenia with fractures secondary to minor or unrecognized trauma, scoliosis, or hyperextensible joints [1, 2, 3, 4]. Dental abnormalities include the retention of primary teeth, the failure of secondary teeth to erupt, and delayed resorption of the roots of the primary teeth [1, 2].
Although this disorder was first noted in Caucasians, it also has been observed in Asians and individuals of African descent [2]. HIE is considered to be an autosomal dominant disorder with incomplete penetrance, so there is appreciable variation in presentation with patients often displaying a partial phenotype. Linkage to chromosome 4q has been identified, but no candidate gene has been found. HIE is characterized by an imbalance between the TH1 and TH2 cytokines, with an increase in the TH2 cytokines IL-4 and IL-13 compared to the TH1 cytokines IL-12 and IFN-γ. The peripheral blood mononuclear cells produce less IL-12 and subsequently IFN-γ compared to normal controls in response to S. aureus but not to other antigenic stimuli [5]. This alteration is important because IL-12 and IFN-γ can be used in vitro to down-regulate the production of IgE by the peripheral blood mononucluear cells of patients with HIE. Other cytokines such as IL-6 and IL-8 play a role in potentiating (IL-6) or blocking (IL-8) the production of IgE in HIE and suggest new targets for therapeutic intervention [6].
Treatment of the dermatitis in HIE is accomplished by the use of topical glucocorticoids and emollients, and the infections are treated with oral antibiotics, wound care, and incision and drainage of abscesses [1, 2, 3, 4]. Other therapies, which include cimetidine, ascorbic acid, cromoglycate, cyclosporine, isotretinoin, intravenous immunoglobulin, plasmapheresis, and IFN-γ, have been limited to case reports, and no controlled trials have been attempted because of the rarity of the disease [2].
References
1. Grimbacher B, et al. Hyper-IgE syndrome with recurrent infections: an autosomal dominant multisystem disorder. N Engl J Med 1999;340:692.2. Erlewyn-Lajeunesse, MDS. Hyperimmunoglobulin-E syndrome with recurrent infection: a review of current opinion and treatment. Pediatr Allergy Immunol 2000;11:133.
3. Borges WG, et al. The face of Job. J Pediatr 1998;133:303.
4. Shemer A, et al. The hyper-IgE syndrome: two cases and review of the literature. Int J Dermatol 2001;40:622.
5. Borges WG, et al. Defective interleukin-12/interferon-? in patients with hyperimmunoglobulinemia E syndrome. J Pediatr 2000;136:176.
6. Garraud O, et al. Regulation of immunoglobulin production in hyper-IgE (Job's) syndrome. J Allergy Clin Immunol 1999;103:333.
© 2004 Dermatology Online Journal