Palmoplantar keratoderma as the initial sign in a peripheral T-cell lymphoma
Published Web Location
https://doi.org/10.5070/D365s6k52gMain Content
Palmoplantar keratoderma as the initial sign in a peripheral T-cell lymphoma
Wendi E Wohltmann MD (Maj USAF MC FS)1, Daniel M MacAlpine MD2, Darryl Shaw Hodson MD3
Dermatology Online Journal 16 (5): 3
1. Wilford Hall Medical Center, San Antonio, Texas. wwohltmann@hotmail.com2. Dermatology, PC, Des Moines, Iowa
3. Skin Surgery Center, Winston-Salem, North Carolina
Abstract
A 27-year-old male with an acquired severe, diffuse palmoplantar keratoderma as the initial sign of peripheral T-cell lymphoma is reported.
Introduction
Palmoplantar keratoderma (PPK) is an inherited or acquired condition characterized by diffuse or localized hyperkeratosis of the palms and soles. The acquired form has been described as a paraneoplastic finding in a variety of malignancies or can develop, as well as a result of direct involvement of the skin by cutaneous T-cell lymphoma (CTCL). We report a case of acquired PPK that preceded the diagnosis of peripheral T-cell lymphoma (PTCL) by 14 months. This report illustrates the importance of acquired PPK as a potential harbinger to PTCL.
Case report
A 27-year-old African-American male was referred to Wilford Hall Medical Center for evaluation of a 14-month history of increasing thickness and roughness of the skin on the palms and soles, along with crumbling of his fingernails and toenails. Two months prior to presentation, generalized lymph node enlargement was noted on a routine physical exam, after which he began experiencing night sweats, fatigue, and episodes of fever and chills. He also had an eight-month history of plaques on his scalp. All other review of systems and past medical history were negative. He denied a history of psoriasis or dyshidrosis. His only treatment up to this point was a trial of a low potency topical corticosteroid to his hands. He had no family history of dermatologic disorders or internal malignancy.
 |  |
| Figure 1 | Figure 2 |
|---|---|
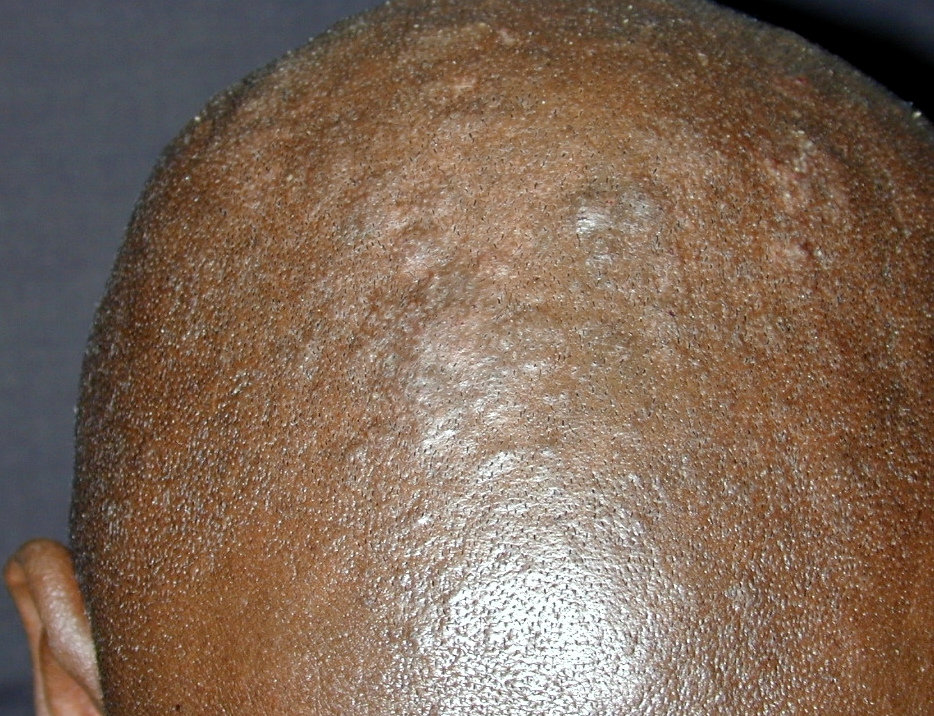
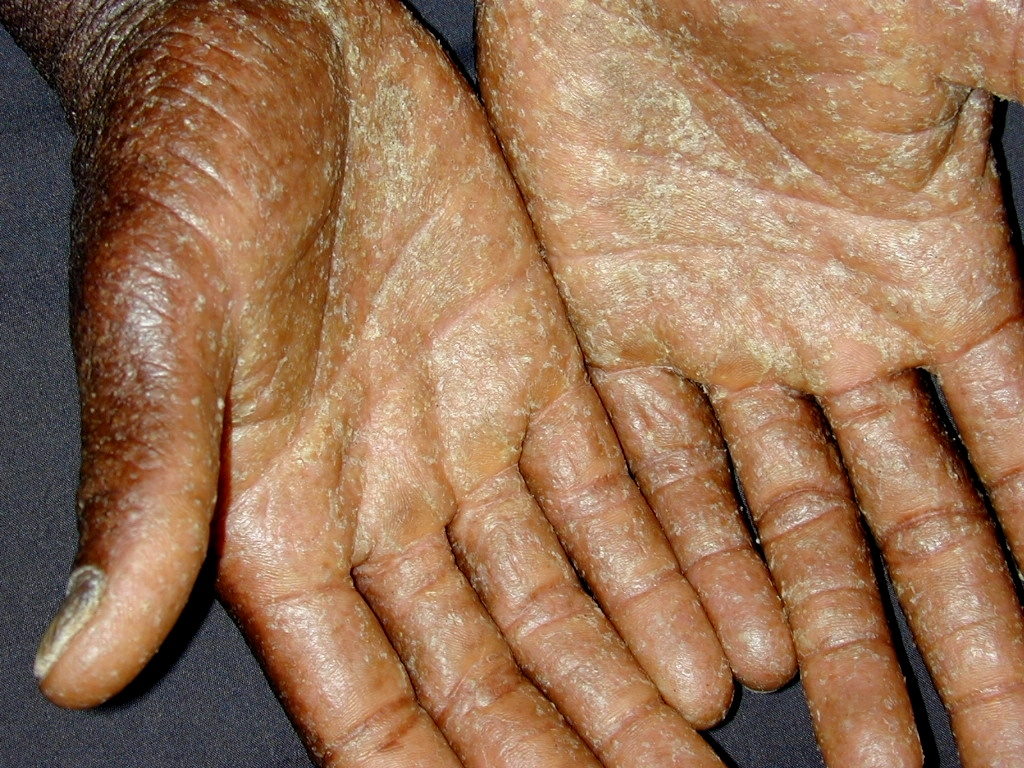
| Figure 1. Large, soft confluent plaques of the scalp Figure 2. Coalescent hyperkeratotic plaques with small pustules on the palms | |
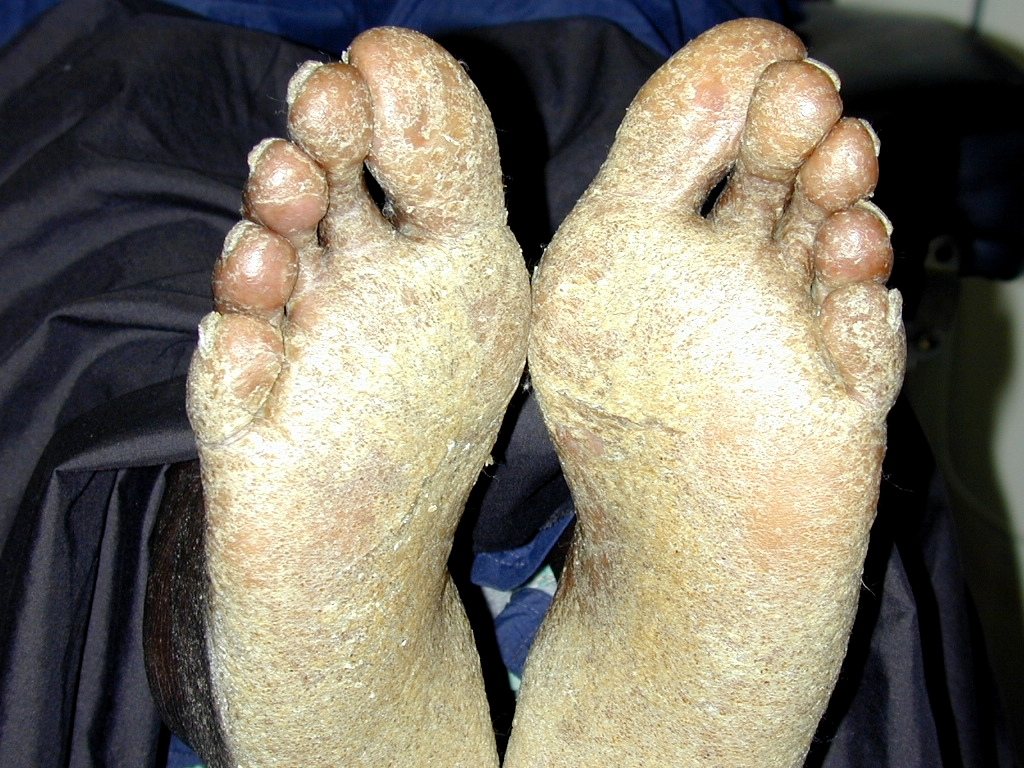
On exam, the patient had large, soft confluent plaques on his scalp (Figure 1). His oropharynx showed violaceous discoloration over the bilateral buccal mucosa. There was shotty cervical and impressive bilateral inguinal lymphadenopathy. His palms and soles had coalescent hyperkeratotic plaques with a few small pustules on his hands (Figures 2 and 3). All of his nails were pitted, thickened, and dystrophic (Figure 4). Also noted was marked xerosis over his trunk and extremities with some subtle patchy hypopigmentation.
 |  |
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Diffuse hyperkeratotis of the soles Figure 4. Pitted, thickened and dystrophic toenails | |
The plaques on his palms and soles were KOH negative. No fluid was expressed from the tiny pustules on his palms. Aspartate aminotransferase was 54 U/L (normal 0 to 37 U/L) and lactate dehydrogenase was 647 U/L (normal 105 to 233 U/L). White blood cell count was 21,000/mm³ (normal 4,800 to 10,800/mm³) with 37 percent atypical lymphocytes; the peripheral smear showed Sezary-like cells. Beta-2-microglobulin level, erythrocyte sedimentation rate, and coagulation lab values were all normal. Antibody tests for hepatitis A, B, and C and human T-lymphotropic virus types 1 and 2 were nonreactive. Examination of the patient’s blood demonstrated a T-cell receptor gamma chain gene rearrangement, indicating evidence of a monoclonal population of T-cells.
Computerized tomography of the chest, abdomen, and pelvis revealed bilateral axillary, inguinal, and iliac chain lymphadenopathy. A positron emission tomography scan showed bilateral axillary uptake and mild diffuse omental uptake.
 |  |
| Figure 5 | Figure 6 |
|---|---|
| Figure 5. Biopsy specimen of the scalp shows cellular atypical lymphoid infiltrates extensively involving the superficial
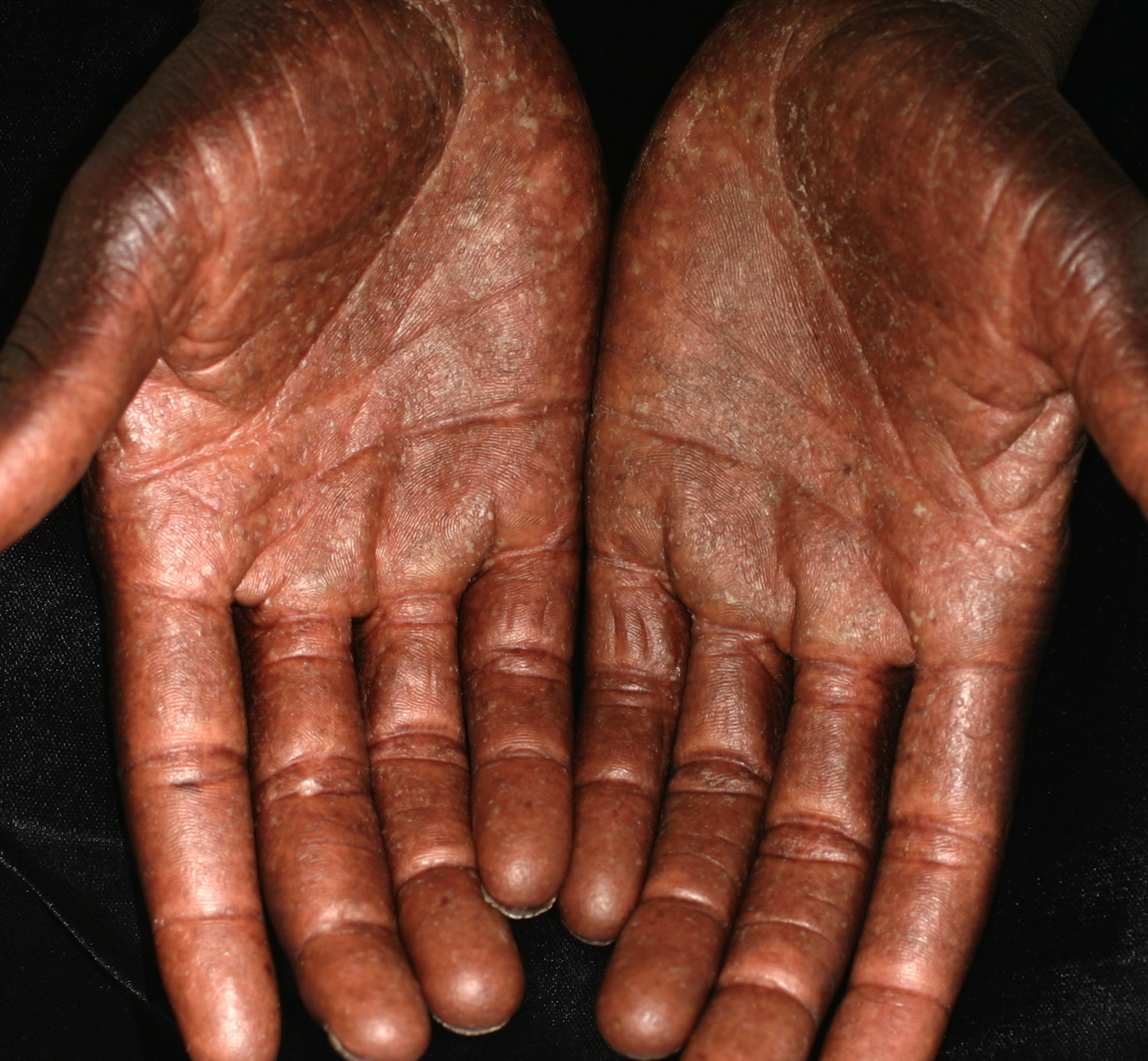
and deep dermis as well as Pautrier-type microabscesses located within the follicular epithelium (H&E, x10) Figure 6. Diffuse thin, hyperkeratotic plaques with scattered pustules of the palms after chemotherapy | |
Two punch biopsies were taken from the scalp. Both showed a dense atypical lymphoid infiltrate containing cells of varying sizes with cleaved or convoluted nuclei extensively involving the superficial and deep dermis, as well as Pautrier-type microabscesses located within the epidermis and follicular epithelium (Figure 5). The neoplastic cells were immunoreactive for CD3, CD4 (weak), CD5, and CD43, with partial loss of CD7 (aberrant immunophenotype). A few cells expressed cytoplasmic and membranous CD30 positivity. CD20 and CD79a highlighted a significant population of accompanying B-cells. Endomysial antibodies and ALK-1 were negative. A biopsy of an axillary lymph node was interpreted as PTCL and an examination of the peripheral blood and bone marrow was shown to be involved by a T-cell lymphoproliferative disorder. The above combined findings, in conjunction with the clinical presentation, led to a diagnosis of stage IV PTCL with cutaneous involvement.
A course of eight cycles of chemotherapy consisting of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) was initiated. A topical regimen of urea 40 percent cream applied to the affected areas each night and clobetasol 0.05 percent ointment twice a day as needed for the hyperkeratosis was started for treatment of the PPK. The generalized xerosis was treated with the daily application of fluocinolone 0.01 percent oil after bathing.
The patient was seen for follow-up six months after the initial presentation. He had received six cycles of CHOP at three-week intervals. The patient stated that after the fourth cycle of CHOP, the PPK resolved for a short time and then flared with each subsequent treatment. He was no longer using topical medications. On physical exam, the scalp lesions, lymphadenopathy, xerosis, and dyspigmentation had all resolved. His palms and soles showed marked improvement with residual diffuse thin, hyperkeratotic plaques with scattered pustules (Figure 6). His nails also showed improvement. After two more cycles of CHOP, the plan is for bone marrow ablation and transplantation.
Comment
In summary we present a 27-year-old male with stage IV PTCL who developed a diffuse PPK over one year prior to diagnosis and six months prior to other cutaneous manifestations. PPK is a condition characterized by diffuse or localized hyperkeratosis of the palms and soles. Clinically, three patterns have been identified: diffuse over the palm and sole, focal with large keratin masses at points of friction, and punctate with tiny drops of keratin on the palmoplantar surfaces. It may be subdivided into inherited forms, acquired forms, or syndromes with PPK as an associated feature. Acquired forms have multiple possible causes as listed in Table 1. Common treatment options for PPK, in addition to treating any underlying conditions, include topical preparations such as corticosteroids, salicylic acid, urea, retinoic acid, calcipotriene, and ammonium lactate. Additional treatment modalities consist of phototherapy, debridement [1], oral retinoids [2], or 5-fluorouracil intravenous infusion [3], although this medication itself can cause a PPK.
The differential diagnosis for the acquired type associated with a malignancy includes paraneoplastic forms or manifestations of mycosis fungoides or lymphoma. Acquired PPK as a paraneoplastic sign has numerous documented malignancy associations ranging from adenocarcinoma of the stomach and esophagus to multiple myeloma [4, 5]. It has been suggested that PPK as a paraneoplastic sign is seen more commonly in lymphomas of T-cell origin [6]. Paraneoplastic PPKs usually regress after treatment of the primary disease. Additionally the literature documents patients with diffuse PPK that were biopsied and showed CTCL [7, 8]. In these cases, diffuse PPK usually accompanies CTCL, Sezary type.
There is an acral variant of mycosis fungoides (MF), which represents 0.6 percent of all MF cases. The Ketron-Goodman variant of mycosis fungoides palmaris et plantaris has a tendency to involve palms and soles. Lesions can spontaneously resolve and recur over many years. The clinical course usually is of long duration without progression. Whereas MF is usually CD4+, acral MF can be CD4+, CD8+, or negative for both CD4 and CD8 [7, 8].
Diffuse PPK was the first clinical sign in our patient, who was later diagnosed with stage IV PTCL with bone marrow, lymph node, and cutaneous involvement. As classified by the World Health Organization criteria, PTCL is a category of predominantly nodal T-cell lymphomas that do not correspond to other more clearly defined subtypes of peripheral T-cell neoplasms such as anaplastic large cell lymphoma, adult T-cell leukemia/lymphoma, angioimmunoblastic T-cell lymphoma, or T-cell rich B-cell lymphoma. Peripheral T-cell lymphoma comprises about half of the peripheral T-cell neoplasms diagnosed in western countries. A form of non-Hodgkins lymphoma, it is a primary monoclonal T-cell lymphoma that usually presents with nodal and peripheral blood involvement. Although any site may be affected, infiltrates are common in the bone marrow, liver, spleen, and skin [9]. This type of lymphoma has an unfavorable prognosis when presenting in the skin, irrespective of the presence or absence of extracutaneous disease at the time of diagnosis. The amount of skin affected has not been shown to correlate with survival rates. Patients presenting with solitary or localized skin lesions have survival rates that are roughly equal to those presenting with multifocal lesions of the skin [10]. Progression of PTCL to stage IV is common. In one study of 36 patients with PTCL diagnosed by the World Health Organization criteria, 69.4 percent had stage IV disease. Complete remission and partial remission were achieved in 12 and 10 out of 31 patients treated with CHOP-based chemotherapy, respectively. One- and two-year overall survivals were 60.6 and 25.0 percent, respectively. Significant negative prognostic factors for survival were found to include elevated serum LDH, diminished performance status, and the presence of fever, night sweats, and weight loss [11]. In a patient presenting with acquired PPK, a basic malignancy work-up might be prudent to rule out any underlying disease, even in the absence of any other symptoms.
Conclusion
In summary, our patient presented with an acquired PPK as the earliest sign of PTCL. New onset PPK should give a high index of suspicion for the diagnosis of malignancy, including both peripheral and cutaneous T-cell lymphoma. After an initial work-up, patients should undergo close follow-up and biopsies if indicated. Acquired PPK may be an important potential harbinger to PTCL.
Abbreviations UsedPPKCTCL
PTCL
KOH
EMA
ALK-1
CHOP
LDH
MF
palmoplantar keratoderma
cutaneous T-cell lymphoma
peripheral T-cell lymphoma
potassium hydroxide
endomysial antibodies
activin receptor-like kinase 1
cyclophosphamide, doxorubicin, vincristine, prednisone
lactate dehydrogenase
mycosis fungoides
References
1. Redmond A, Allen N, Vernon W. Effect of scalpel debridement on the pain associated with plantar hyperkeratosis. J Am Podiatr Med Assoc. 1999 Oct;89(10):515-519. [PubMed]2. Capella GL, Fracchiolla C, Frigerio E, Altomare G. A controlled study of comparative efficacy of oral retinoids and topical betamethasone/salicylic acid for chronic hyperkeratotic palmoplantar dermatitis. J Dermatolog Treat. 2004 Apr;15(2):88-93. [PubMed]
3. Lienemann AO, Colucci VJ, Jones MS, Trauscht JM. Treatment of palmoplantar keratoderma with continuous infusion 5-fluorouracil. Cutis. 2004 May;73(5):303-308. [PubMed]
4. Murata I, Ogami Y, Nagai Y, Furumi K, Yoshikawa I, Otsuki M. Carcinoma of the stomach with hyperkeratosis palmaris et plantaris and acanthosis of the esophagus. Amer J Gastroenterology. 1998 Mar;93(3):449-451. [PubMed]
5. Smith CH, Barker JN, Hay RJ. Diffuse plane xanthomatosis and acquired palmoplantar keratoderma in association with myeloma. Br J Dermatol. 1995 Feb;132(2):286-289. [PubMed]
6. Trattner A, Katzenelson V, Sandbank M. Palmoplantar keratoderma in a noncutaneous T-cell lymphoma. Int J Dermatol. 1991 Dec;30(12):870-871. [PubMed]
7. Aram H, Zeidenbaum M. Palmoplantar hyperkeratosis in mycosis fungoides. J Am Acad Dermatol. 1985 Nov;13(5 Pt 2):897-899. [PubMed]
8. Resnik KS, Kantor GR, Lessin SR, Kadin ME, Chooback L, Cooper HS, et al. Mycosis fungoides palmaris et plantaris. Arch Dermatol. 1995 Sep;131(9):1052-6. [PubMed]
9. Jaffe ES, Harris NL, Stein H, Vardiman JW. WHO Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, Lyon. 2001;216-220;227-229.
10. Bekkenk MW, Vermeer MH, Jansen PM, van Marion AMW, Canninga-van Dijk MR, Kluin PM, et al. Peripheral T-cell lymphomas unspecified presenting in the skin: analysis of prognostic factors in a group of 82 patients. Blood. 2003 Sep 15;102(6):2213-2219. Epub 2003 May 15. [PubMed]
11. Kojima H, Hasegawa Y, Suzukawa K, Mukai HY, Kaneko S, Kobayashi T, et al. Clinicopathological features and prognostic factors of Japanese patients with "peripheral T-cell lymphoma, unspecified" diagnosed according to the WHO classification. Leuk Res. 2004 Dec;28(12):1287-1292. [PubMed]
© 2010 Dermatology Online Journal