Epidermolysis bullosa acquisita
Published Web Location
https://doi.org/10.5070/D36222v7f3Main Content
Epidermolysis bullosa acquisita
Jennifer A Stein MD PhD, Radha Mikkilineni MD
Dermatology Online Journal 13 (1): 15
New York University Department of DermatologyAbstract
A 29-year-old woman presented with 8 months of multiple vesicles, erosions, and milia on the dorsa of her hands and feet. Histopathologic examination demonstrated a subepidermal blister, with a paucity of eosinophils and a lack of blood vessel wall thickening or caterpillar bodies. A direct immunofluorescence test showed a linear deposit of IgG at the dermo-epidermal junction. These findings were consistent with a diagnosis of epidermolysis bullosa acquisita. This case is a classic example of this rare blistering disease, in which patients produce autoantibodies to collagen VII, which is the major component of the anchoring fibrils.
Clinical synopsis
A 29-year-old woman presented to the Dermatology Clinic at Bellevue Hospital Center for evaluation of painful blisters and erosions on her hands and feet, which had started 8 months prior to her initial visit. She had not been treating the lesions. The patient has mitral valve prolapse and congenital deafness. Medications include a multivitamin and iron. There is no family history of similar lesions. A biopsy specimen confirmed the diagnosis, and treatment was initiated with dapsone 50 mg daily.
Multiple, 3-5 mm vesicles and erosions were present on the dorsa of the hands as well as on the thenar eminences and feet. Several milia also were noted on the hands. No oral lesions were evident.
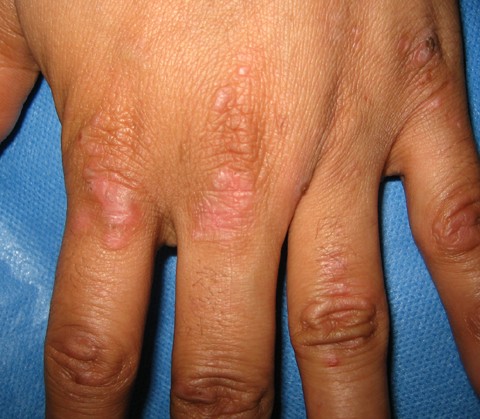 |  |
| Figure 1 | Figure 2 |
|---|---|
A complete blood count and chemistry and hepatic profiles were normal. Antinuclear antibody was negative.
Histopathology reveals a subepidermal bulla with fibrin and a paucity of inflammatory cells. Adjacent to this, there is a small milial cyst. A direct immunofluorescence test showed linear deposits of IgG at the dermo-epidermal junction.
Comment
Epidermolysis bullosa acquisita (EBA) is an autoimmune blistering disease that is mediated by antibodies against collagen VII, which is the major component of the anchoring fibrils in the lamina densa of the basement membrane [1]. The disease is rare, with an estimated annual incidence of 0.25 per million in Western Europe [2]. It classically presents in the third-to-fifth decade with scarring blisters and milia at sites of trauma, such as the dorsa of the hands and feet as well as the elbows, knees, and buttocks [3]. Other subsets of patients also may have mucosal involvement that can mimic cicatricial pemphigoid or can present with inflammatory blisters that mimic bullous pemphigoid. The blisters of the classic mechanobullous non-inflammatory EBA patients resemble those observed in dystrophic epidermolysis bullosa, which is not surprising based on the involvement of collagen VII in both diseases [4].
Most EBA patients have IgG antibodies that recognize the non-collagenous domain of collagen VII, although there is a subset of patients with IgG that targets the collagenous domain. There is also a subset of patients with IgA, rather than IgG autoantibodies [5]. Evidence for a causative relationship between the presence of collagen VII antibodies and blister formation comes from the observation that passive transfer of antibodies from EBA patients to mice is able to cause blister formation in areas of trauma in the mouse. The fact that blister formation does not occur in mice lacking proper complement activation ability suggests that the mechanism of EBA blister formation is complement mediated [6, 7, 8]. Complement deposition is observed along with immunoglobulins at the basement membrane in EBA patients [4].
Clinically, EBA can resemble bullous pemphigoid, porphyria cutanea tarda, or bullous lupus erythematosus. The diagnosis of EBA requires both clinical and pathologic evidence. On histopathologic examination, a subepidermal blister is observed, and a direct immunofluorescence test shows IgG in a linear array along the dermoepidermal junction. Other confirmatory tests include an indirect immunofluorescence test to show the presence of circulating antibodies in the serum with the ability to target collagen VII [4]. Salt-split skin will show decoration of the base of the vesicle with immune deposits [9]. This test can help to differentiate EBA from bullous pemphigoid, in which immune complexes decorate the roof of the vesicle. EBA also lacks the abundant eosinophils seen in bullous pemphigoid. EBA can look strikingly similar to porphyria cutanea tarda, but the presence of autoantibodies at the DE junction and the lack of blood vessel wall thickening and caterpillar bodies helps to distinguish it. Immunoelectron microscopy, which shows autoantibodies binding to the anchoring fibrils, is the gold standard of diagnosis of EBA [4].
EBA patients generally have a normal lifespan, although the disease can be particularly difficult to treat. It is often resistant to a variety of medications that include systemic glucocorticoids. Colchicine, dapsone, azathioprine, gold, intravenous immunoglobulins, mycophenolate mofetil, and cyclosporin have been successful in some patients [4, 10]. A recent case of successful treatment with rituximab has been reported [11]. Care is generally directed at avoidance of trauma and wound care for the blisters and erosions [4].
References
1. Woodley D, et al. Identification of the skin basement membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med 1984;310: 10072. Bernard P, et al. Incidence and distribution of subepidermal autoimmune bullous diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995;131:48
3. Gammon WR, et al. Epidermolysis bullosa acquisita - a pemphigoid-like disease. J Am Acad Dermatol. 1984, 11: 820
4. Borradori L, Bernard P, Pemphigoid Group. In: Bolognia J, et al., eds. Dermatology. London: Mosby, 2003: 463
5. Vodegel RM, et al. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. 2002;47:919
6. Borradori L, et al. Passive transfer of autoantibodies from a patient with mutilating epidermolysis bullosa acquisita induces specific alterations in the skin of neonatal mice. Arch Dermatol 1995;131:590
7. Sitaru C, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest 2005;115:870
8. Woodley D, et al. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol 2005;124:958
9. Gammon WR, et al. Differentiating anti-lamina lucida and anti-sublamina densa anti-basement membrane zone antibodies by indirect immunofluoresence on 1.0 M sodium chloride separated skin. J Invest Dermatol 1984;82:139
10. Fine JD Management of acquired bullous skin diseases. N Engl J Med 1995;333:1475
11. Schmidt E, et al. Successful adjuvant treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody rituximab. Arch Dermatol 2006;142:147
© 2007 Dermatology Online Journal