Type I hereditary punctate keratoderma
Published Web Location
https://doi.org/10.5070/D35hz345n9Main Content
Type I hereditary punctate keratoderma
Arash K Asadi MD
Dermatology Online Journal 9(4): 38
From the Ronald O. Perelman Department of Dermatology, New York University
Abstract
A 75-year-old man with a history of prostatic carcinoma and atypical fibroxanthoma reports a long-standing history of 1-2 mm depressed, hyperkeratotic papules on the palms. His mother suffered a similar condition. Histologically, the papules demonstrated hyperkeratosis, without columns of parakeratosis. A diagnosis of type I hereditary punctate keratoderma (Buschke-Fisher-Brauer disease) was made. This condition, which is classified as one of the three hereditary forms of punctate palmoplantar keratoderma, is an autosomal-dominant condition with variable penetrance. It is characterized clinically by multiple, tiny, punctate keratoses of the palms and soles. Affected individuals appear to be at increased risk of developing malignant conditions.
Clinical summary
History.—A 75-year-old man presented for a many-year problem of papules on the palms. The patient has a long history of asymptomatic lesions on the palm, which has become more apparent after immersion in water. Past medical history includes hypertension, gout, and prostate cancer. Medications include diltiazem and lisinopril. His dermatologic history includes an atypical fibroxanthoma, which was excised in August 2000, and numerous solar keratoses, which were treated with 5-fluorouracil and liquid nitrogen cryotherapy. The patient reported that his mother had similar lesions on the palms.
Physical examination.—On the palms were numerous 1-2 mm depressed, hyperkeratotic papules. There were no punctate lesions on the soles, but there was focal hyperkeratosis that affected the pressure points.
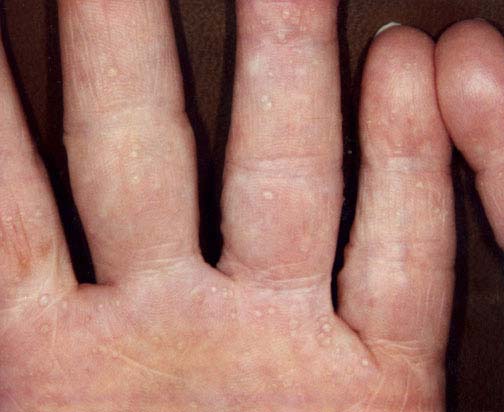
|
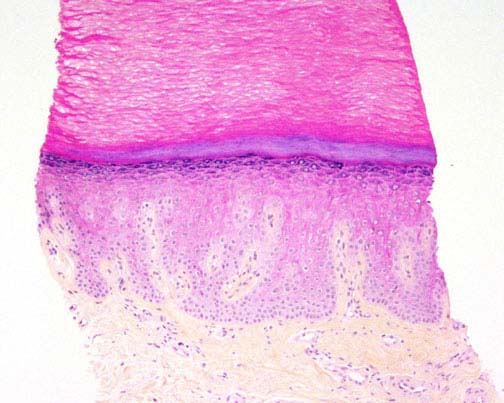
|
| Figure 1 | Figure 2 |
|---|
Radiologic and laboratory data.—A chest radiograph and computerized tomography scan showed a left lung nodule. A colonoscopy performed in November 2002 was normal. The prostate specific antigen was normal.
Histopathology.—There is epidermal hyperplasia with hypergranulosis and marked orthokeratotic hyperkeratosis. There is a superficial, perivascular infiltrate of lymphocytes.
Diagnosis.—Type I hereditary punctate keratoderma.
Comment
The four categories of palmoplantar keratoderma (PPKs) include diffuse, focal, and punctate types as well as the palmoplantar ectodermal dysplasias. The punctate PPKs are subdivided into acquired and hereditary forms. Acquired forms include arsenical keratoses, which are associated with angiosarcoma of the liver, nonmelanoma skin cancer, and bronchial adenocarcinoma; idiopathic punctate PPK, which is associated with increasing age and internal malignant conditions; idiopathic filiform porokeratotic PPK, which is associated with breast, renal, colon, and lung cancer; and punctate PPK of the palmar creases, which occurs predominantly in Afro-Caribbeans and in association with an atopic diathesis.
The hereditary forms of punctate PPKs were classified into three types: Type I (Buschke-Fisher-Brauer disease, keratosis punctata, and keratosis papulosa) is an autosomal-dominant condition with onset between ages 12 and 30. Multiple tiny, punctate keratoses are found, and there is a possible association with malignant conditions. Type II (porokeratosis punctata, palmaris, et plantaris) is an autosomal-dominant condition with onset between ages 12 and 50. There are numerous, tiny, keratotic spines, which resemble the spines of a music box. Histologically, these spines correspond to columnar parakeratosis that resemble cornoid lamellae. Type III (acrokeratoelastoidosis lichenoides) is an autosomal dominant condition. There are oval or polygonal crateriform papules on the borders of the hands, feet, and wrists as well as in the center of the palms and soles. No gene has yet been identified for hereditary punctate PPKs, and linkage to chromosomes 12 and 17 has been excluded in some pedigrees [2].
Features that support the diagnosis of Type I hereditary punctate PPK in this patient include a family history that supports autosoma-dominant transmission; clinical findings of numerous tiny, punctate keratoses; and absence of a coronoid lamella histologically. A four-generation pedigree has been reported that comprises over 320 individuals, of whom 49 had punctate PPK [3]. The inheritance pattern was autosomal dominant with variable penetrance. The incidence of malignant conditions in this pedigree was 23 percent in adults affected with punctate PPK as compared to 2 percent in unaffected adults. Associated malignant conditions included Hodgkin disease as well as renal, breast, pancreatic, and colon adenocarcinomas. This patient's prostatic carcinoma and atypical fibroxanthoma are likely sporadic, although his punctate PPK may have served as a risk factor for the development of these malignant conditions.
References
1. Stevens HP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24: literature survey and proposed updated classification of the keratodermas. Arch Dermatol 132:640, 1996.2. Kelsell DP, et al. Genetic linkage studies in non-epidermolytic palmoplantar keratoderma: evidence of heterogeneity. Hum Mol Genet 4:1021, 1995.
3. Stevens HP, et al. Punctate palmoplantar keratoderma and malignancy in a four-generation family. Br J Dermatol 134:720, 1996.
© 2003 Dermatology Online Journal