Dyschromatosis universalis hereditaria in an African American male
Published Web Location
https://doi.org/10.5070/D35f89b1z7Main Content
Dyschromatosis universalis hereditaria in an African American male
Shruthi Geedipalley Reddy MD, Sophie Marie Worobec MD
Dermatology Online Journal 17 (8): 3
Department of Dermatology, University of Illinois at Chicago, Chicago, IllinoisAbstract
Dyschromatosis universalis hereditaria (DUH) is a very rare genodermatosis characterized by generalized skin dyspigmentation. It is most common in Japan, but has also been reported in other parts of Asia, Europe, South America, and Africa. We report a case of a 44-year-old man born and raised in North America who presented with total skin discoloration since birth.
Introduction
Dyschromatosis universalis hereditaria (DUH), which has been subtyped into DUH 1 [Online Mendelian Inheritance in Man (OMIM) 127500], an autosomal dominant form, and DUH 2 [OMIM 612715] an autosomal recessive form, is a rare genodermatosis characterized by generalized hypopigmented and hyperpigmented macules of varying sizes and shapes occurring all over the body. Dyschromatosis universalis hereditaria was first described in 1933 by Ichikawa and Higari [1] and is most commonly reported in Japan [2]. However, DUH has also been reported in other parts of Asia, Europe, South America, and Africa [3-9]. We present a case of DUH in an African American male.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|
 |
| Figure 3 |
|---|
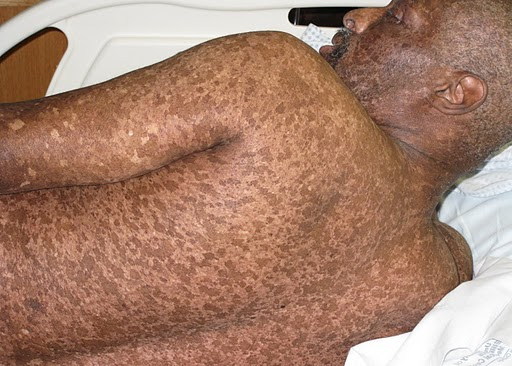
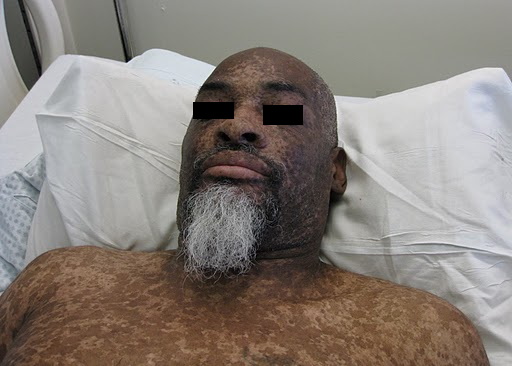
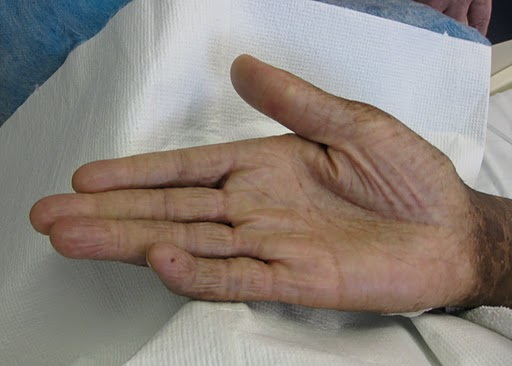
A 44-year-old man, born and raised in North America, presented with generalized hypopigmented and hyperpigmented macules and patches of varying shades, shapes, and sizes sparing the palms, soles, and oral mucosa (Figures 1, 2, and 3). No poikiloderma, atrophy, or telangiectasias were present. The dyspigmentation was noted at birth and stable since childhood. The patient reported mild pruritus, but was otherwise asymptomatic. He recalled being evaluated at age 14 by a dermatologist and undergoing multiple skin biopsies. However he did not recall a diagnosis. The patient denied a family history of skin discoloration or other skin conditions. His past medical history was significant for Crohn disease.
 |
| Figure 4 |
|---|
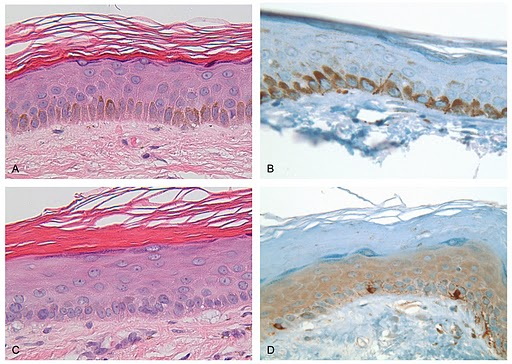
Histological examination of a hyperpigmented macule showed increased melanin deposition within the keratinocytes with a normal number and distribution of melanocytes highlighted by both H&E and Melan-A staining (Figure 4, A and B). In contrast, histological examination of a hypopigmented macule showed a reduced amount of melanin within the keratinocytes in the lesional area with no change in the number of melanocytes (Figure 4, C and D).
Discussion
Dyschromatoses are a diverse group of disorders characterized by hyperpigmented and hypopigmented macules. Classification of hereditary dyschromatoses depends on the distribution and extent of skin lesions and includes: the generalized forms, DUH 1 and DUH 2, a localized form limited to the extremities, dyschromatosis symmetrica hereditaria [OMIM 127400], also called acropigmentation of Dohi, and a segmental form, unilateral dermatomal pigmentary dermatosis. Familial progressive hyperpigmentation (FPH) [OMIM 145250] is an autosomal dominantly inherited disorder characterized by hyperpigmented patches that present in early infancy and progress with age. The absence of hypopigmented lesions differentiates FPH from DUH [10]. Generalized reticulate pigmentary dermatoses include dermatopathia pigmentosa reticularis characterized also by nail dystrophy, nonscarring alopecia of the scalp, eyebrows and axilla, absent dermatoglyphics, and melanin pigmentation of the optic fundi; Naegeli-Franceschetti-Jadassohn syndrome, an autosomal dominant syndrome characterized by ectodermal dysplasia affecting the sweat glands, nails, teeth, and skin with hypoplastic dermal ridges, keratoderma of the palms and soles with punctate keratosis, and dental anomalies; and dyskeratosis congenita characterized by the triad of reticulate pigmentation of the skin, nail dystrophy with failure to form a nail plate, and leukokeratosis of the oral mucosa [7]. Other conditions characterized by cutaneous dyschromia include xeroderma pigmentosum, chronic radiodermatitis, amyloidosis, malnutrition, pigmentary changes secondary to topical application of chemicals such as diphencyclopropenone or monobenzyl ether of hydroquinone, photoleukomelanodermatitis related to medications, and acquired brachial cutaneous dyschromatosis [5], and post-inflammatory hypo- and hyperpigmentation from a variety of dermatoses or skin injuries.
Dyschromatosis universalis hereditaria is characterized by generalized asymptomatic hypo- and hyperpigmented macules of varying sizes and shapes. Eighty percent of affected individuals develop dyschromia before age 6 and approximately 20 percent present at birth with dyschromia. There is a slight female preponderance. The dyschromia may rarely involve the palms and soles but usually spares the mucous membranes [6, 7, 8]. Isolated reports have illustrated mucous membrane involvement, including the tongue [8]. Fifty percent of affected individuals demonstrate facial lesions [5]. No seasonal change or spontaneous regression with age has been reported. Systemic abnormalities are rare and include short stature, high-frequency deafness, erythrocyte, platelet and tryptophan metabolism abnormalities, insulin-dependent diabetes mellitus, bilateral glaucoma and unilateral cataract, photosensitivity along with neurosensory hearing defects, and grand-mal seizures [7]. Dyschromatosis universalis hereditaria has also been reported in association with other conditions such as Dowling-Degos disease, X-linked ocular albinism, and tuberous sclerosis [8].
In 1992, Kim et al. [11] examined both histologic and ultrastructural findings of both hyperchromic and achromic macules from a 43-year-old man with DUH. Fontana-Masson stained hyperchromic macules showed heavily stained melanin pigments in the basal layer of the epidermis with melanophages in the upper dermis. In contrast, achromic macules showed no melanin pigmentation and clear basal cells, which appeared to be inactive melanocytes. With electron microscopic examination, hyperchromic macules showed an increased number of fully melanized melanosomes forming melanosome complexes in the keratinocytes, but these melanosomes were absent from both keratinocytes and melanocytes in the achromic areas. The authors therefore concluded that DUH appears to be a disorder of melanosome production rather than a disorder of melanocyte number [11], with normal numbers of morphogenetically intact and active melanocytes [4, 7]. On H&E, our patient's biopsies showed normal number and distribution of melanocytes and increased amount of melanin in hyperpigmented skin and reduced amount of melanin in hypopigmented skin (Figure 4). In addition, DUH may be related to the interference of the neural reflex-melanocyte interaction early in embryonic life in genetically susceptible individuals [5].
In 1952, Suenega [2] reported a Japanese family with DUH, in which a consanguineous marriage occurred in 4 successive generations, which suggested an autosomal recessive inheritance pattern. Bukhari et al. in 2006 [9] reported a Bedouin family with evidence of autosomal recessive transmission. Nuber in 2004 [4] reported a family in which there were two cases of father to son transmission, compatible with autosomal dominant transmission. Thus, the reported inheritance pattern of DUH is variable with autosomal dominant mode described in the majority of cases and additional reports of sporadic, autosomal recessive, and pseudodominant transmission [2, 3, 6, 8, 9]. Gene loci responsible have been mapped to chromosome 12 in one autosomal recessive form (DUH 2 [OMIM 612715]) [9, 12], and to chromosome 6q24.2-q25.2 in an autosomal dominant form (DUH 1 [OMIM 127500]), which was labeled DSH in the reporting paper by Xing et al. [13]. However, because the clinical description of the affected individuals described dyschromatosis of the trunk, face, neck and arms, this was thought by Miyamura et al. [14] to actually represent DUH with linkage to 6q24.2-q25.2. Thus far, the gene candidates for chromosome 6 or 12 have not been definitively identified, making the diagnosis of DUH rely mainly on external phenotype [8, 9, 12, 13]. The most promising gene for the region on chromosome 12 is the pro-melanin-concentrating hormone (PMCH) gene. PMCH has been suggested to play a role in regulating skin pigmentation [12].
To the best of our knowledge, we report the first case of DUH in North America. This conclusion was based on a detailed PubMed and Medline search using the search terms dyschromatosis universalis hereditaria, dyschromatosis, universalis, and hereditaria. In addition, images in medical, dermatological, and melanocyte biology textbooks were searched for descriptions of DUH affecting patients found in North America and none were found. Although more commonly reported in Asia than in Europe and Africa, we would like to remind clinicians that DUH can be seen in North America and the diagnosis should be kept in mind for patients presenting with diffuse mottled dyspigmentation.
References
1. Ichigawa T, Hiraga Y. A previously undescribed anomaly of pigmentation dyschromatosis universalis hereditaria. Jpn J Dermatol Urol 1933; 34:360-364 (in Japanese).2. Suenaga, M. Genetical studies on skin diseases. VII. Dyschromatosis universalis hereditaria in five generations. Tohoku J. Exp. Med. 55: 373-376, 1952. [PubMed]
3. Yusuf SM, Mijinyawa MS, Maiyaki MB, and Mohammed AZ. Dyschromatosis universalis hereditaria in a young Nigerian female. Int J Dermatol 2009; 48(7):749-750(2). [PubMed]
4. Nuber UA, Tinschert S, Mundlos S, and Haußer I. Dyschromatosis universalis hereditaria: Familial case and ultrastructural skin investigation. Am J Med Genet A 2004;125(3):261-266. [PubMed]
5. Al Hawsawi K, Al Aboud K, Ramesh V, and Al Aboud D. Dyschromatosis universalis hereditaria: report of a case and review of the literature. Pediatr Dermatol 2002; 19:523-526. [PubMed]
6. Kenani N, Ghariani N, Denguezli M, Sriha B, Belajouza C, and Nouira R. Dyschromatosis universalis hereditaria: Two cases. Dermatol Online J 2008 Feb 28; 14(2):16. [PubMed]
7. Sethuraman G, D'Souza M, Thappa DM, Srinivas CR, and Smiles L. Dyschromatosis universalis hereditaria. Clin Exp Dermatol 2002; 27:477-479. [PubMed]
8. Udayashankar C and Nath, AK. Dyschromatosis universalis hereditaria: A case report. Dermatol Online Journal 2011 Feb 15; 17(2): 2. [PubMed]
9. Bukhari IA, El-Harith EA, and Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. Europ. Acad. Derm. Venerol. 18: 628-629, 2006. [PubMed]
10. Wang ZQ, Si L, Tang Q, Lin D, Fu Z, Zhang J, Cui B, Zhu Y, Kong X, Deng M, Xia Y, Xu H, Le W, Hu L, Kong X. Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet. 2009 May;84(5):672-7. [PubMed]
11. Kim NS, Im S, Kim SC. Dyschromatosis universalis hereditaria: an electron microscopic examination. J Dermatol. 1997 Mar;24(3):161-4. [PubMed]
12. Stuhrmann M, Hennies HC, Bukhari IA, Brakensiek K, Nürnberg G, Becker C, Huebener J, Miranda MC, Frye-Boukhriss H, Knothe S, Schmidtke J, and El-Harith EH. Dyschromatosis universalis hereditaria: evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin Genet 2008; 73:566-572. [PubMed]
13. Xing Q, Wang M, Chen X, Feng G, Ji H, Yang J, Gao J, Qin W, Qian X, Wu S, and He L. A gene locus responsible for dyschromatosis symmetrica hereditaria (DSH) maps to chromosome 6q24.2-q25.2. Am J Hum Genet. 73: 377-382, 2003. [PubMed]
14. Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, and Tomita Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 73: 693-699, 2003. [PubMed]
© 2011 Dermatology Online Journal