Lipoid Proteinosis in a 12-year-old child: a report from west India
Published Web Location
https://doi.org/10.5070/D353p9q04qMain Content
Lipoid Proteinosis in a 12-yea-old child: a report from west India.
Dr Sangeeta Kini MD1, Dr Ashok Jain MD2, Dr Tanuja M Shet MD3, Dr Sangeeta Bansode MD 4, Dr Ila M Vora MD1, Dr Kanchanmala Ghorpade MD1
Dermatology Online Journal 12 (1): 10
1.Department of Pathology, Terna Medical College and Hospital, Navimumbai. sangukini@yahoo.co.in 2. Department of Dermatology, Terna Medical College and Hospital, Navimumbai.
3. Assistant Pathologist, Department of Pathology, Tata Memorial Hospital, Parel, Mumbai.
4.Lecturer, Department of Pathology, Navimumbai Municipal Medical Corporation, Navimumbai
Abstract
A 12-year-old male child born of non-consanguineous parents presented with multiple skin lesions, hoarseness of voice, and episodes of epilepsy since early childhood. The findings of characteristic beaded eyelid margins, patchy alopecia of the scalp, hoarseness of voice, and epilepsy were consistent with a rare clinical diagnosis, lipoid proteinosis. Skin biopsies obtained from representative skin lesions were subjected to histology and electron microscopy. Light microscopy demonstrated PAS-positive diastase-resistant material in the papillary dermis of skin. Ultrastructure revealed granulo-filamentary aspect of the accumulated material. Although this rare autosomal recessive disorder has been described in the literature, its occurrence is rare in India.
Introduction
Lipoid proteinosis (LP) also known as hyalinosis cutis et mucosae is a rare, recessively-inherited form of congenital and hereditary disorder described by Urbach and Weithe in 1929 [1]. It is characterized by protean clinical manifestations attributed to deposition of homogenous hyaline-like material in dermis and viscera. The exact pathogenesis and chemical composition of the material in LP remains unknown. However, profound alterations of extracellular membrane protein 1 (EMP1) confirmed by molecular methods [2] and disturbances of collagen metabolism confirmed by biochemical, immunological, and ultrastructural methods have been found to be associated with LP [3, 4, 5]. Occurrence of lipoid proteinosis has been consistently described in the literature. We report this case of lipid proteinosis from west India as this entity still remains rare in Indian literature.
Clinical synopsis
A 12-year-old male child born of nonconsanguineous parents presented to us with multiple asymptomatic variable raised skin lesions present since early childhood. The patient had a normal outcome at birth but started developing hoarseness of voice and frequent epileptic episodes during infancy followed by crops of bullae and pustules which healed with atrophic scars. There was no history of photosensitivity.
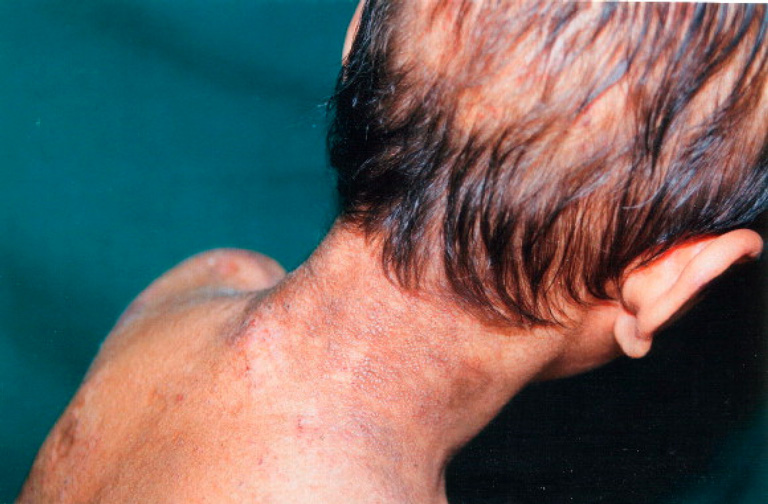
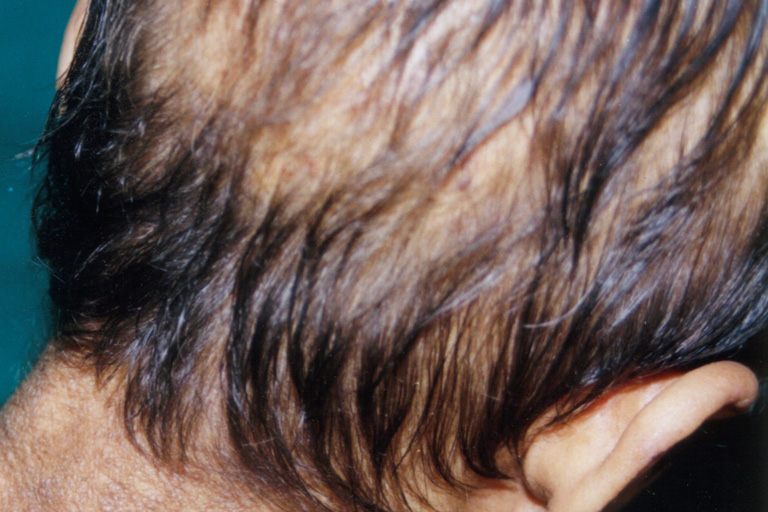
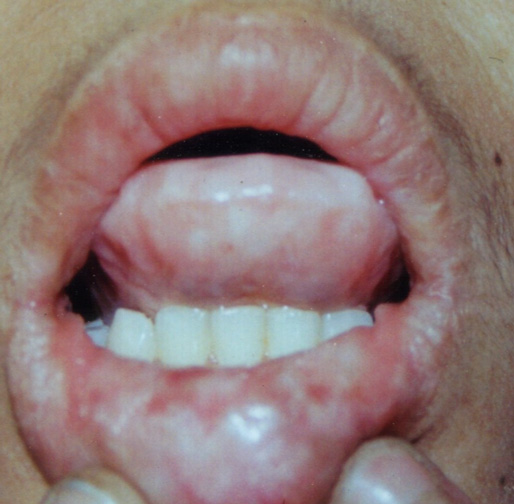
On examination, the patient had hoarseness of voice and numerous skin-colored waxy papules on the face, forehead, and nape of neck. Patchy areas of alopecia were widespread on the scalp (Figs. 1 and 2). Infiltrative papular deposits were seen on the inner surface of lips, undersurface of tongue, fauces, and uvula. The tongue was large, thickened, infiltrated and showed teeth indentations at the margins (attributed to inability to protrude the tongue)(Fig. 3). Dentition was normal.
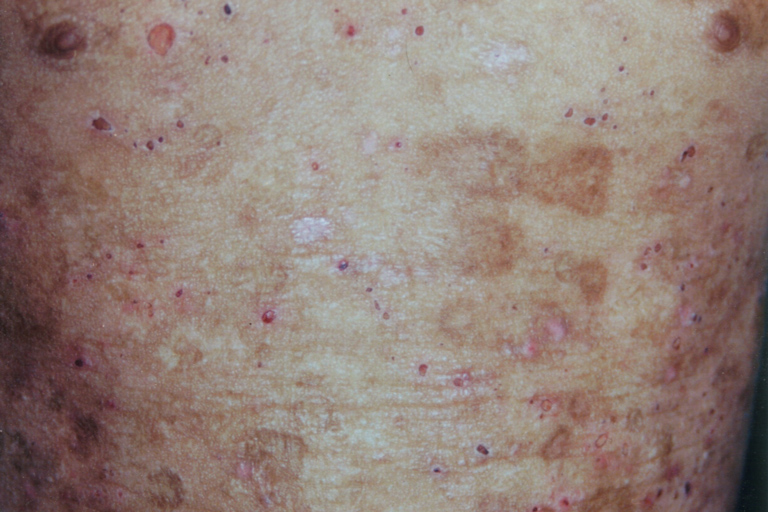
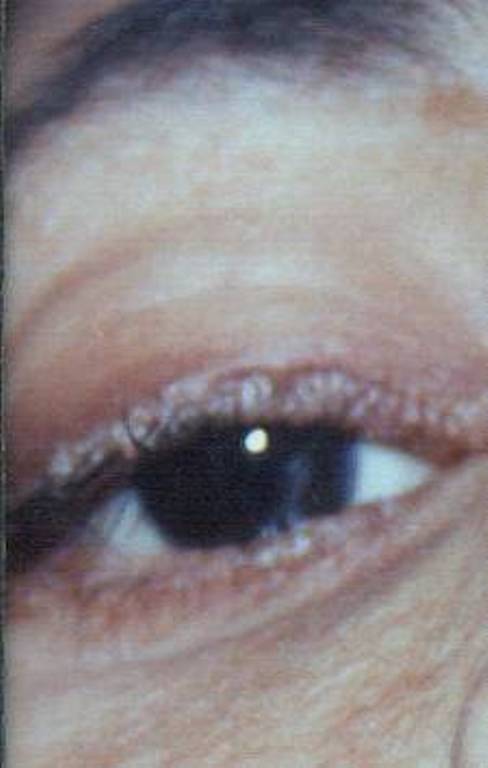
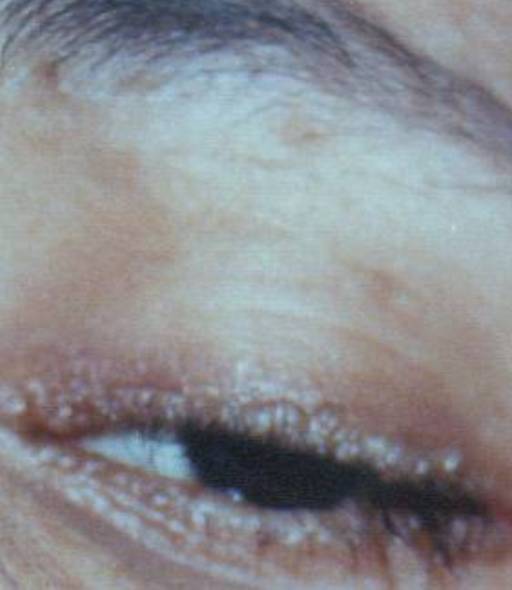
Hyperkeratotic wart-like lesions were seen on the elbows and knees. The trunk showed healed atrophic scars (Fig. 4). Pearly beaded papules were seen at both the upper and lower eyelid margins (so-called moniliform blepharosis). Fundus examination was normal (Figs. 5 and 6)
The rest of the systemic examination was found to be normal.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Waxy papular eruptions on the nape of the neck with pock-like marks. | |
| Figure 2. Patchy alopecia seen all over the scalp. | |
 |  |
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Infiltrative papular deposits on the inner surface of the lips and woody indurated tongue with teeth indentations at its margins. | |
| Figure 4. Atrophic pock-like scars over the trunk. | |
 |  |
| Figure 5 | Figure 6 |
|---|---|
| Figures 5 and 6. Beaded papules on both eyelid margins. | |
An MRI of the brain done during early childhood in workup of epilepsy showed flattening of the skull vault in the right frontal region and with shallow right hemicranium and consequent entire impression on right hemispheric lobe, which may be post-traumatic or consequent to premature sutural fusion. No focal parenchymal abnormality was noticed. Routine hematological and biochemical investigations were within normal limits.
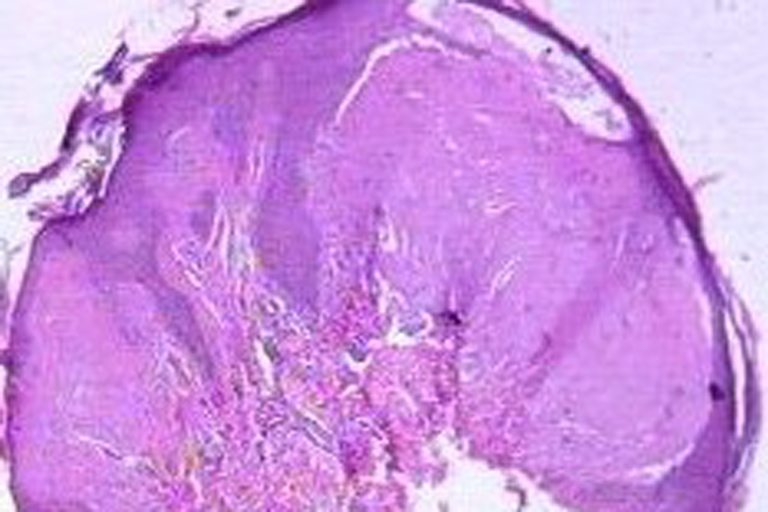
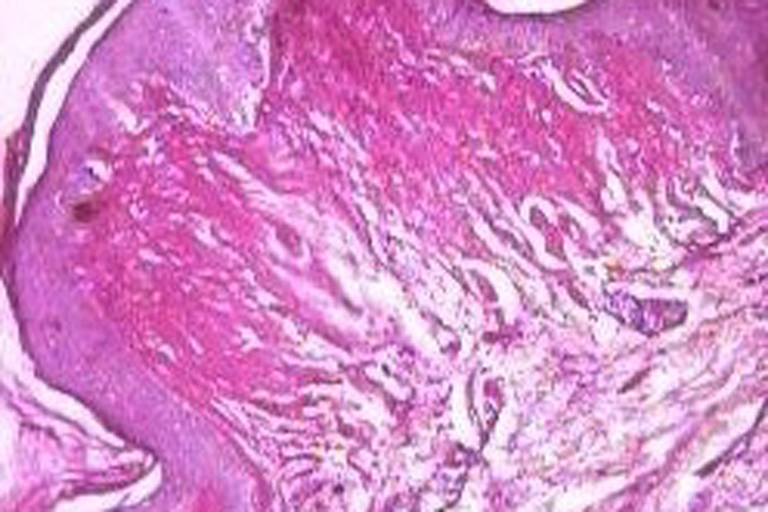
Histopathological examination of representative skin lesions revealed abundant deposition of PAS positive diastase resistant eosinophilic hyaline-like material in the papillary dermis, surrounding the blood vessels and adnexal structures (Figs. 7 and 8).
 |  |
| Figure 7 | Figure 8 |
|---|---|
| Figures 7 and 8. Acellular PAS positive diastase resistant eosinophilic hyaline like material is seen deposited in the dermis surrounding the blood vessels and appendages (H & E and PAS with diastase 40x). | |
 |
| Figure 9 |
|---|
| Figure 9. Focal epidermal thinning and thick homogenous eosinophilic bundles perpendicular to the overlying epidermis are seen (H&E 40x). |
Focal epidermal thinning was present above the deposited material and the deposited material had thick slender prolongation perpendicular to the skin surface (Fig. 9).
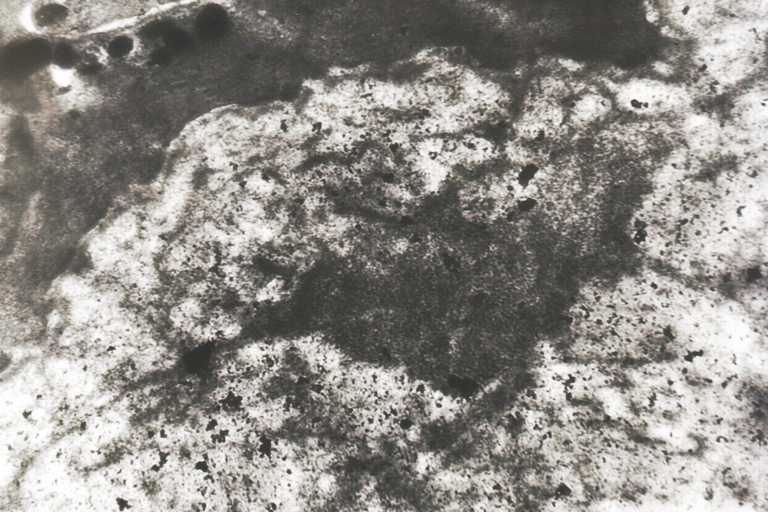
Ultrastructural examination of the skin biopsy specimen fixed in 2 percent gluteraldehyde revealed granulo-filamentary aspect of the accumulated material next to the epidermal cells (Fig. 10) and multilamination of the basal laminae at the dermo-epidermal junction (Fig. 11).
 |  |
| Figure 10 | Figure 11 |
|---|---|
| Figure 10 & Figure 11. Granular-filamentary aspect of the accumulated material next to the epidermal cells & multilamination of the basal laminae. | |
Discussion
Lipoid proteinosis is a rare recessively-inherited disorder characterized by deposition of eosinophilic hyaline-like material in skin and viscera, leading to protean clinical manifestations [2]. The classic clinical features such as eyelid beading, thickened woody tongue, hoarseness of voice, variable skin lesions (such as waxy papules, warty lesions, and scars), and epileptic episodes are typical for the diagnosis of LP. Extracutaneous manifestations such as epilepsy and neuropsychiatric illnesses are less commonly seen [6]. There was no similar clinical background found in the siblings of this patient as described in the literature [7, 8].
LPis clinically differentiated from erythropoietic protoporphyria (EPP) by absence of photosensitivity and presence of skin lesions in non-sun exposed areas. Histologically PAS positive material is differentiated from amyloid by negative staining for amyloid on Congo red stain.
Treatment is usually limited and variable results have been found with Dimethylsulphoxide, D-penicillamine, and etretinate. Our patient is managed on conservative lines with multi-vitamin and anti-epileptic drugs and routine follow-up on out patient basis has showed no further progressive lesions currently.
We report this case because of its rarity and because of its association with the less common clinical finding of epilepsy with lipoid proteinosis.
Acknowledgment: We thank Department of Pathology, Tata Memorial Hospital, Parel, Mumbai to perform the electron microscopy of the skin biopsy specimen of the said case.
References
1. FabiriG, Perfiri B, Borgioli M, Serri F, Urbach-Weithe disease: Light and Electron microscopic study, J Cutan Pathol. 7:8-20,1980. PubMed.2. Hammada T, Lipoid proteinosis, Clin Exp Dermatol. Nov 27(8):624-29, 2002. PubMed.
3. Moy LS, Moy RL, Matsouka LY, Ohta A, Uitto J, Lipoid proteinosis: Ultrastructural and Biochemical studies, J Am Acad Dermatol. !6(6):1193-1201, 1987. PubMed.
4. Navaroo C, Fachal L, Rodriguez C, Padro L, Dominguez C, Lipoid proteinosis: A biochemical and ultrastructural investigation of two new cases, Br J Dermatol. 141:326-331, 1999. PubMed.
5. Newton JA, Rasbridge S, Temple A, Pope Fm, Black MM, Mckee P, Lipoid proteinosis: new immunopathological observations, Cin Exp Dermatol. Sep 16(5):350-354, 1991. PubMed.
6. Kleinert R, Cervos- Navarro J, Kleinert G, Walter GF, Steiner H, Predominantly cerebral manifestations in Urbach-Weithe's syndrome (lipoid proteinosis cutis et mucosae): a clinical and pathological study, Clin Neuropathol. Jan-Feb 6(1):43-45, 1987. PubMed.
7. Shivaswamy KN, Thappa DM, Laxmisha C, Jayanthi S, Lipoid proteinosis in two siblings :a report from south India, Dermatol Online J. Dec 9(5):12, 2003. PubMed.
8. Vedmurthy M, Lipoid proteinosis in siblings. Dermatol Online J. Dec 9(5):13, 2003. PubMed.
© 2006 Dermatology Online Journal