Heterogeneity and atypical presentation in infantile systemic hyalinosis with severe labio-gingival
Published Web Location
https://doi.org/10.5070/D353h877w4Main Content
Heterogeneity and atypical presentation in infantile systemic hyalinosis with severe labio-gingival enlargement: First Egyptian
report
Ghada Y El-Kamah1, Mostafa I Mostafa2
Dermatology Online Journal 15 (5): 6
1. Clinical Genetics Department, Division of Human Genetics and Genome Research National Research Centre, Cairo, Egypt.2. Oro-dental Genetics Department, Division of Oral and Dental Medicine Research National Research Centre, Cairo, Egypt. mostafanrc@yahoo.com
Abstract
Infantile systemic hyalinosis (ISH) (MIM 236490) is a rare, progressive, fatal autosomal recessive condition characterized by widespread deposition of hyaline material in many tissues. Our proband was a 4-year-old male with growth retardation, severe labio-gingival enlargement, generalized stiff skin, joint contractures, and intractable diarrhea. We discovered a history of a brother and sister who suffered a more severe disease course. A final diagnosis of systemic hyalinosis was made; we report this case and discuss the clinical and orodental heterogeneity among these siblings in the first report of an Egyptian family with ISH. We present a very rare entity, infantile systemic hyalinosis, a cause of joint contracture, protein-losing enteropathy, and growth retardation in infancy with a review of the relevant literature.
Introduction
Infantile systemic hyalinosis (ISH) (MIM 236490) [1] is a rare, progressive, fatal autosomal recessive condition [2]. Until 1994, 11 patients with ISH had been reported [3] and all of them died by early childhood, mainly due to severe diarrhea, pulmonary infections, or septicemia. Since then sporadic cases have been reported. In the available international English literature the last report in 2009 was by Al-Mubarak et al. [4].
Infantile systemic hyalinosis is characterized by widespread deposition of hyaline material in many tissues such as skin, gastrointestinal tract, adrenals, skeletal muscles, gingiva and other loci [5, 6]. Clinical features include painful swollen joint contractures, hyperpigmentation over bony prominences, diffuse thickening of the skin with pearly papules (predominantly of the face, scalp, and neck) and fleshy nodules (particularly in the perianal region), osteoporosis of bones, bone fractures, short stature, persistent diarrhea, and failure to thrive [1, 4, 7].
Children with ISH are intellectually normal. However, the symptoms become apparent soon after birth and death usually occurs before the age of two years [6, 7, 8]. If they survive infancy, patients become less susceptible to infection and their joints may become less painful. However, their mobility remains severely restricted by joint contractures [1]. Histologically, ISH is characterized by widespread hyaline deposition [9, 10]. Molecular analysis has revealed mutations in the capillary morphogenesis protein 2 (CMG2) gene as the cause of both ISH and juvenile hyaline fibromatosis (JHF) [11]; additional CMG2 mutations continue to be reported [12]. Herein we study an atypical presentation and discuss the clinical and orodental heterogeneity among the studied family members.
Clinical presentation
 |  |
| Figure 1 | Figure 2 |
|---|---|
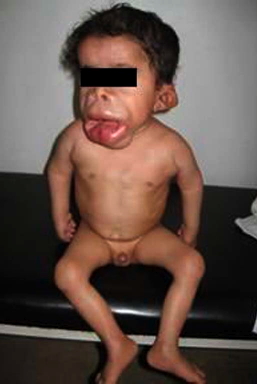
| Figure 1. Proband's dysmorphic features & multiple confluent papules at the lips, chin & ala nasi. Tight skin and joint contractures
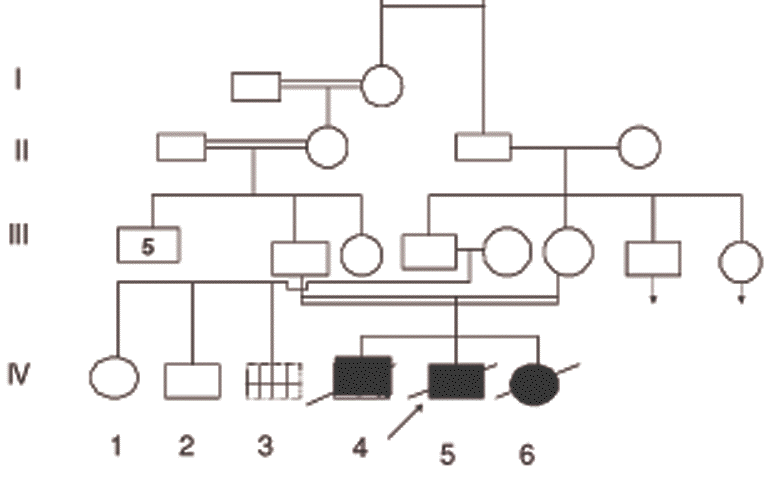
specially shown at the elbows & hands. Figure 2. Pedigree of the studied family (case IV-3 had mental retardation, no acquired motor skills but there was a history suggestive of obstructive labor) | |
A 4-year-old boy (IV-5) (Fig. 1) presented at our clinic with marked labio-gingival enlargement and painful joint contractures in knees, elbows, shoulders, and pelvis. He was normal until the age of 4 months when his parents noticed that he experienced discomfort on being handled and showed some difficulties in moving his limbs. At the age of one year, his parents started to notice papules on his nose, perioral area, ears, and perianal area in addition to increased head size with no weight gain. He had no history of seizures or hoarseness, but suffered episodes of respiratory infection and diarrhea.
He was born at full term by a normal vaginal delivery following an uneventful pregnancy. His mother and father are second degree, healthy consanguineous, 23 and 26 year-olds, respectively. The parents, in turn, descend from a closed consanguineous pedigree. Our patient had delayed milestones and was unable to stand. Mental development could not be assessed because the patient was distressed from the large swellings obstructing the oral cavity.
Our patient's birth was preceded by a male (IV-4) who died at the age of three days because of respiratory distress and a sister (IV-6) who died at the age of 21 months from repeated attacks of intractable diarrhea. She had painful flexural contractures of the elbows, wrists and knees, which started at about the age of 4 months. The pedigree is shown in Figure 2.
Examination of our index case showed that he was ill and pale with abnormal, coarse facies including anteverted nose, and low set ears as part of his craniofacial dysmorphism. He was short for age (height about 80 cm, sitting height 53.5, -5.5SD). His weight was 12 kg (-2.8SD) and his head circumference was 55.5 (+3SD). He was responsive to his surroundings but irritated and could not support himself in a standing position.
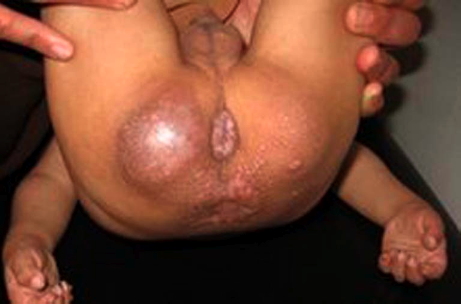
Cutaneous examination revealed the presence of multiple, skin-colored and pink papules of variable sizes over his ear lobes, inside his ear, around his nose, and on the nasolabial folds, chin, and mouth. They were clustered together and confluent at the lips, ear lobules, and chin. The trunk and limbs were spared. He had multiple, pearly, grouped papules on the dorsum of the neck and in the perianal region (Fig. 3). In addition to rectal prolapse, early pressure ulcers were noted on his buttocks. Overall, the skin was not thickened, but showed hyperpigmented areas over the metacarpophalangeal joints and the malleoli.
 |  |
| Figure 3 | Figure 4 |
|---|---|
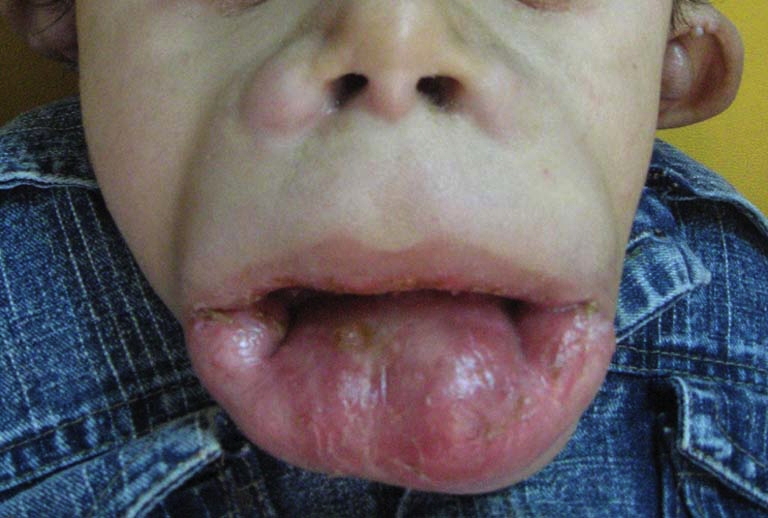
| Figure 3. Periananl papules & rectal prolapse. Figure 4. Long philtrum, open mouth, severe labial enlargement and the lower lip is severely everted. | |
 |  |
| Figure 5 | Figure 6 |
|---|---|
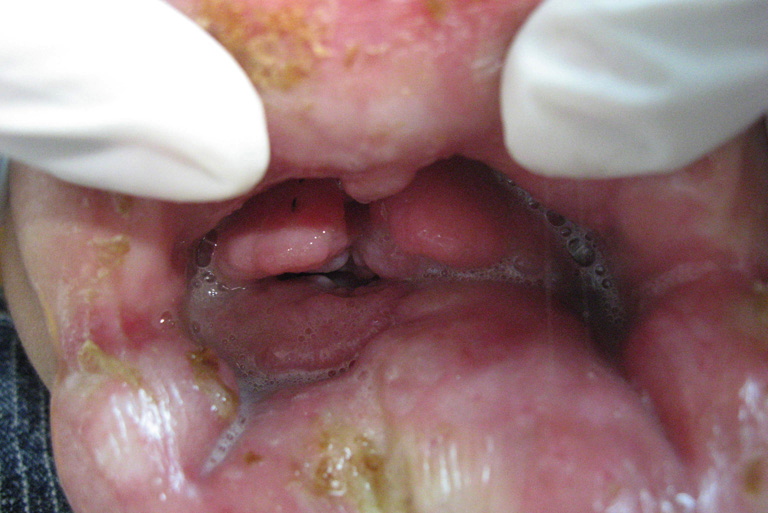
| Figure 5. Severe gingival enlargement, Both lips are crusted, the gingival tissue is firm, normal pink color, not ulcerated
or covered with pseudomembranes. The gingival tissue almost covered all erupting teeth. Figure 6. Multiple sclerosing erosions in the proximal femora. | |
Oro-dental examination (Figs. 4 & 5) revealed a long philtrum, open mouth, and severe labio-gingival enlargement that partially occluded the oral cavity, making feeding very difficult. Both lips were firm and crusted; the lower lip was severely everted. The gingival tissue was firm, a normal pink color and without easy bleeding, ulceration or pseudomembrane. The gingival tissue almost covered all erupting teeth and was obstructing his breathing.
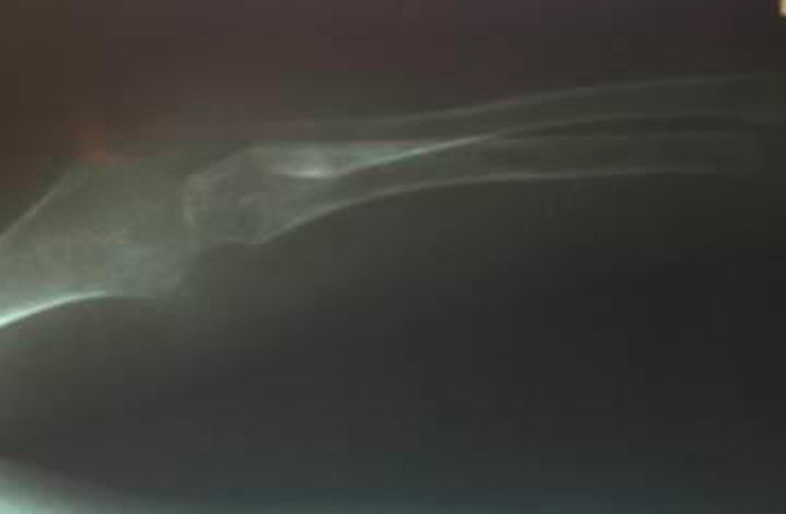
Complete blood count, renal function tests, liver function tests, blood sugar, and electrolytes were within normal limits. Chest X-ray was normal. Skeletal radiographs revealed a wide anterior fontanel, thick vertebral bodies, and diffusely demineralized long bones with multiple sclerotic erosions in the parietal bones of the calvarium, ends of both clavicles, proximal humeral metaphyses, and the neck and capital epiphyses of the proximal femoral and tibial metaphyses (Figs. 6 & 7). Non-contrast CT brain scan (with bone window settings) revealed atrophic brain changes; widened cortical sulci, silvian fissure, basal cistern ventricular system, and a prominent gyral pattern. Echocardiography revealed a normal heart with pulmonary hypertension, (PAP 45mm Hg) secondary to upper airway obstruction.
 |  |
| Figure 7 | Figure 8 |
|---|---|
| Figure 7. Bony erosions affecting the long bones of the arm. Figure 8. Hyalinosis in dermal papillae (x200) | |
A skin biopsy of the papules showed a normal epidermis with deposits of amorphous, eosinophilic hyaline material throughout the upper and mid dermis (Fig. 8).
The child was diagnosed as a case of infantile systemic hyalinosis. Although we had not examined his brother and sister, the features as per his mother's history (especially for the sister) are suggestive of a severe and rapidly fatal form of ISH (Table 1). The parents were counseled about the progressive nature of the disease and the 25 percent chance of development of disease in future offspring.
Discussion
Infantile systemic hyalinosis (infantile hyaline fibromatosis, infantile hyalinoses) shares many clinical similarities to juvenile hyaline fibromatosis (JHF) [13]. ISH & JHF are both caused by mutations in the CMG2 gene [11]. However, the pathogenesis of hyalinosis syndromes is unclear. Ultra-structurally an apparent increase in the amount of collagen type VI was suggested to account for the clinical features of inflexible skin and the limitation of joint movement in ISH [13,14]. ISH has been mostly linked to mutations in the von Willebrandt factor A (vWA) domains in the CMG2G gene. In recent studies of the cellular consequences of vWA mutations, they were mostly found to cause retention of protein in the endoplasmic reticulum (ER), through different mechanisms. Studies suggested that in vivo, slow folding, rather than misfolding, is responsible for ER retention; systemic hyalinosis can be considered to be a conformational disease, at least for the mutations that have been mapped to the extracellular and transmembrane domains [15].
The main clinical features of both ISH and JHF include papular and nodular skin lesions, gingival hyperplasia, join contractures, and diverse degrees of bone involvement [16]. Distinguishing features attributed to ISH include diffuse, thickened skin, hyperpigmented plaques on bony prominences, visceral involvement, persistent diarrhea, frequent severe infections, and fatal outcome [17]. Almost all of those distinguishing features were found proband and his sister (Table 1). Because presentation is usually at birth or within the first few months of life [18], case IV-4 could have exhibited a severe neonatal form with very early death before developing the rest of disease manifestations.
Osteopenia is often present and results in increased susceptibility to bone fractures. Severe joint limitation and pain lead to immobility and respiratory insufficiency. Feeding problems, malnutrition, and protein-losing enteropathy are caused by the thickening and hyaline infiltration of the intestinal walls. Death occurs secondary to sepsis with renal, respiratory and heart failure, usually by the age of two years. [7, 10, 19] Case IV-4 died at the age of 3 days from severe respiratory failure and case IV-6 died at the age of 21 months from recurrent infections and intractable diarrhea. However, case IV-5 atypically survived until the age of 4½ years and died as a complication of anesthesia.
One ISH patient with atypical prolonged survival has been reported, but survival was until the age of 3 years. [20]. Other reports have discussed the difficulty in distinguishing ISH from JHF [7, 19, 21, 22]. It has been pointed out that the criterion of survival should no longer be used in differentiating ISH and JHF because improvement in clinical care may allow for prolonged life expectancy [10].
Both ISH and JHF are allelic disorders resulting from mutations in CMG2 [7] found on chromosome 4q21. [11]. Both conditions are linked to a disorder of the synthesis of glycosaminoglycans with resulting abnormalities in collagen synthesis [23].
Reviewing the literature, no genotype variations were reported among siblings. However, the clinical picture of our studied cases pointed out the presence of a significantly heterogeneous phenotypic expression among family members. Case IV-4 died at the age of 3 days, case IV-6 had a severe disease course, and case IV-5 lived beyond double the expected lifespan for ISH. In addition to the prolonged survival, our index case exhibited atrophic brain changes and severe gingival hypertrophy to a greater degree than described in previous reports. It is possible that his increased lifespan allowed these developments. Both manifestations were not present in other affected family members. Heterogeneity was also noted in some JHF families in the study conducted by Hanks et al. 2003 [24].
Histologically, ISH is characterized by hyaline deposition that can affect several tissues [9, 10]. Hyaline deposits in the skin are noted in other disorders such as amyloidosis and lipoid proteinosis. Perivascular hyaline deposits are a feature of porphyria, especially erythropoietic protoporphyria [7, 25].
In our case amyloidosis was not evident by histopathological studies [26]; he did not have hoarseness of voice, or atrophic skin scarring. His biopsy did not show the deposition of amorphous, eosinophilic, hyaline material within the dermis with an onion skin arrangement around blood vessels, characteristic of lipoid proteinosis. His laboratory investigations, disease history, and pathological studies were inconsistent with porphyria [25].
Different degrees of gingival enlargement could be considered a common feature of some disorders, such as neurofibromatosis, gingival hyperplasia, congenital generalized myofibromatosis, and Winchester syndrome [27]. Inborn metabolic disorders such as Farber disease, mucopolysaccharidoses, ligneous periodontitis, and I-cell disease should also be considered in differential diagnosis [27, 28].
Our index case had cutaneous lesions typical for hyalinosis and not reported with neurofibromatosis or gingival fibromatosis. He had no corneal clouding characteristic of Winchester syndrome and inborn errors of metabolism were excluded on the basis of histological and biochemical findings [23]. The presence or absence of pseudomembrane may also help in differentiation from ligneous periodontitis [8, 28]
He could be differentiated from Farber's disease by the absence of hoarseness of voice, painful nodular lesions, and the characteristic foamy appearance in histopathologic examination [29, 30].
No specific treatment is available for ISH. Early surgical excision is recommended by some authors for those lesions that either present a significant cosmetic problem or produce some degree of functional impairment. However, excision may be followed by recurrences [31]. Spontaneous regression has been reported in some cases, but long-term regression is unlikely [32] and tumors continue to increase in size and number. Intralesional steroid injection may reduce the size of early lesions. Capsulotomy of joints may show some temporary, beneficial effect [33]. Gingival overgrowth may be treated with partial gingivectomy. Oral D-penicillamine has been used in some cases with apparent improvement in joint mobility and flexibility. However, enlargement of some subcutaneous tumors was not inhibited [19].
In conclusion, we present a case of the very rare entity of infantile systemic hyalinosis, a cause of protein-losing enteropathy and growth retardation during infancy. We present a family with clinical heterogeneity and atypical manifestations, even among siblings. Furthermore, our index case died from complications of anesthesia. Special precautions during anesthesia in these patients are of utmost importance [34].
References
1. McKusick VA. Online Mendelian Inheritance in Man (OMIM). Available at: http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim. Accessed, 2009.2. Büyükgebiz B, Öztürk Y, Arslan N, Özer E. A rare cause of protein-losing enteropathy and growth retardation in infancy: infantile systemic hyalinosis. Turk J Pediatr. 2003 Jul-Sep;45(3):258-60. [PubMed]
3. Miyake I, Tokumaru H, Sugino H et al. Juvenile hyaline fibromatosis. Case report with five years' follow-up. Am J Dermatopathol. 1995 Dec;17(6):584-90. [PubMed]
4. Al-Mubarak L, Al-Makadma A, Al-Khenaizan S. Infantile systemic hyalinosis presenting as intractable infantile diarrhea. Eur J Pediatr. 2009 Mar;168(3):363-5. [PubMed]
5. Landing BH, Nadorra R. Infantile systemic hyalinosis: report of four cases of a disease, fatal in infancy, apparently different from juvenile systemic hyalinosis. Pediatr Pediatr Pathol. 1986;6(1):55-79. [PubMed]
6. Glover MT, Lake BD, Altherton DJ. Infantile systemic hyalinosis: newly recognized disorder of collogen?. Pediatrics. 1991 Feb;87(2):228-34. [PubMed]
7. Dhingra M, Amladi S, Savant S, Nayak C. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: Divergent expressions of the same genetic defect?. Indian J Dermatol Venereol Leprol. 2008 Jul-Aug;74(4):371-4. [PubMed]
8. Devlin H, Sloan P, Thakkar NS. Oral manifestations of infantile systemic hyalinosis. J Oral Pathol Med. 1995 Mar;24(3):140-3. [PubMed]
9. Landing BH and Nadorra R. Infantile systemic hyalinosis: report of four cases of a disease, fatal in infancy, apparently different from juvenile systemic hyalinosis. Pediatr Pediatr Pathol. 1986;6(1):55-79. [PubMed]
10. Stucki U, Spycher MA, Eich G, Rossi A, Sacher P, Steinmann B, Superti-Furga A. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature. Am J Med Genet. 2001 Apr 22;100(2):122-9. [PubMed]
11. Rahman N, DunstanM, TeareMD,Hanks S, Edkins SJ,Hughes J, Bignell GR, Mancini G, Kleijer W, Campbell M, Keser G, Black C, Williams N, Arbour L, Warman M, Superti-Furga A, Futreal, PA, Pope FM. The gene for juvenile hyaline fibromatosis maps to chromosome 4q21. Am J Hum Genet. 2002 Oct;71(4):975-80. [PubMed]
12. Lindvall LE, Kormeili T, Chen E, Ramirez MC, Grum-Tokars V, Glucksman MJ, Martignetti JA, Zaragoza MV, Dyson SW. Infantile systemic hyalinosis: Case report and review of the literature. J Am Acad Dermatol. 2008 Feb;58(2):303-7. [PubMed]
13. Huang YC, Xiao YY, Zheng YH, Jang W, Yang YL, Zhu XJ. Infantile systemic hyalinosis: a case report and mutation analysis in a Chinese infant. Br J Dermatol. 2007 Mar;156(3):602-4. [PubMed]
14. Glover MT, Lake BD, Atherton DJ. Clinical, histologic, and ultrastructural findings in two cases of infantile systemic hyalinosis. Pediatr Pediatr Dermatol. 1992 Sep;9(3):255-8. [PubMed]
15. Deuquet J, Abrami L, Difeo A, Ramirez MC, Martignetti JA, van der Goot FG. Systemic hyalinosis mutations in the CMG2 ectodomain leading to loss of function through retention in the endoplasmic reticulum. Hum Mutat. 2009 Apr;30(4):583-9. [PubMed]
16. Mancini GMS, Stojanov L, Willemsen R, et al. Juvenile hyaline fibromatosis: clinical heterogeneity in three patients. Dermatology. 1999;198(1):18-25. [PubMed]
17. Muniz ML, Lobo AZC, Machado MC, Valente NYS, Kim CA, Lourenco S, Nico MMS. Exuberant Juvenile Hyaline Fibromatosis in Two Patients. Pediatr Dermatol. 2006 Sep-Oct;23(5):458-64. [PubMed]
18. Shin HT, Paller A, Hoganson G, Willner JP, Chang MW, Orlow SJ. Infantile systemic hyalinosis. J Am Acad Dermatol. 2004 Feb;50(2 Suppl):S61-4. [PubMed]
19. Urbina F, Sazunic I, Murray G. Infantile Systemic Hyalinosis or Juvenile Hyaline Fibromatosis?. Pediatr Dermatol. 2004 Mar-Apr;21(2):154-9. [PubMed]
20. Thauvin-Robinet C, Faivre L, Beer F, Justrabo E, Nivelon-Chevalier A, Huet F. Infantile systemic hyalinosis: a case with atypical prolonged survival. Acta Paediatr. 2001 Jun;90(6):705-6. [PubMed]
21. Antaya, Richard J, Cajaiba, Mariana, Madri, Joseph, Lopez, Maria A, Ramirez, Maria Celeste M, Martignetti, John A, Reyes-Múgica, Miguel. Juvenile Hyaline Fibromatosis and Infantile Systemic Hyalinosis Overlap Associated With a Novel Mutation in Capillary Morphogenesis Protein-2 Gene. Am J Dermatopathol. 2007 Feb;29(1):99-103. [PubMed]
22. Nofal A, Sanad M, Assaf M, Nofal E, Nassar A, Almokadem S, Attwa E, Elmosalamy K. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: A unifying term and a proposed grading system. J Am Acad Dermatol. 2009 Apr 1. [Epub ahead of print] [PubMed]
23. Mancini G, Orange A, Hollander J, Levy M. Fibromatosis, hyalinosis and Stiff Skin syndrome. In: Harper J, Oranje A, Prose N, editors. Textbook of Pediatric Dermatology. 2nd ed. Oxford: Blackwell 2006; 951-4.
24. Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, Bode H, Campbell ME, Feingold M, Keser G, Kleijer W, Mancini G, McGrath JA, Muntoni F, Nanda A, Teare MD, Warman M, Pope FM, Superti-Furga A, Futreal PA, Rahman N. Mutations in the Gene Encoding Capillary Morphogenesis Protein 2 Cause Juvenile Hyaline Fibromatosis and Infantile Systemic Hyalinosis. Am J Hum Genet. 2003 Oct;73(4):791-800. [PubMed]
25. Weedon D. Skin Pathology. 2nd Ed. Philadephia: Elsevier; 2002; 928.
26. Günhan O, Celasun B, Perrini F, et al: Generalized gingival enlargement due to accumulation of amyloid-like material. J Oral Pathol Med. 1994 Oct;23(9):423-8. [PubMed]
27. Gupta LK, Singhi MK, Bansal M, Khullar R, Jain V, Kachhawa D. Juvenile hyaline fibromatosis in siblings. Indian J Dermatol Venereol Leprol. 2005 Mar-Apr;71(2):115-8. [PubMed]
28. El Darouti M, Zayed A, El Kamah Gh, Mostafa M. Ligneous Conjunctivitis with Oral Mucous Membrane Involvement and Decreased Plasminogen Level. Pediatr Dermatol. 2009 (in press.)
29. Chanoki M, Ishii M, Fukai K, Kobayashi H, Hamada T, Murakami K, Tanaka A. Farber's lipogranulomatosis in siblings: light and electron microscopic studies. Br J Dermatol. 1989 Dec;121(6):779-85. [PubMed]
30. El-Kamah GhY , El-Darouti MA, Kotoury AIS, Mostafa IM. Farber disease syndrome verses stiff skin: Expanding the spectrum. Egypt. J. Med. Hum. Genet. 2009; 10 (1): 135-142. (in press).
31. Finlay AY, Ferguson SD, Holt PJ. Juvenile hyaline fibromatosis. Br J Dermatol. 1983 May;108(5):609-16. [PubMed]
32. Remberger K, Krieg T, Kunze D, Weinmann H-M, Hübner G. Fibromatosis hyalinica multiplex (juvenile hyaline fibromatosis). Light microscopic, electron microscopic, immunohistochemical, and biochemical findings. Cancer. 1985 Aug 1;56(3):614-24. [PubMed]
33. Quintal D, Jackson R. Juvenile hyaline fibromatosis: A 15-year follow-up. Arch Dermatol. 1985 Aug;121(8):1062-3. [PubMed]
34. Pollard M, Ollite EM, Walker RW. The anesthesia management of a child with infantile systemic hyalinosis. Paediatr Anaesth. 2008 Nov;18(11):1123-4. Epub 2008 Jul 29. [PubMed]
© 2009 Dermatology Online Journal