Progressive late-onset of cutaneous angiomatosis as possible sign of cerebral cavernous malformations

Main Content
Progressive late-onset of cutaneous angiomatosis as possible sign of cerebral cavernous malformations
E Campione1, L Diluvio1, A Terrinoni3, A Di Stefani2, A Orlandi2, S Chimenti1, L Bianchi1
Dermatology Online Journal 19 (2): 2
1. Department of Dermatology, University of Rome Tor Vergata2. Institute of Anatomic Pathology, University of Rome Tor Vergata
3. Department of Experimental Medicine, University of Rome Tor Vergata IDI/IRCSS
Abstract
BACKGROUND: Cerebral cavernous malformations (CCM) comprise enlarged capillary cavities in the central nervous system, with possible retinal or cutaneous vascular malformations. This condition is associated with CCM1, CCM2, and CCM3 gene mutations. OBJECTIVE: Cutaneous clinical, histological and cerebral MRI findings, including CCM1, CCM2, and CCM3 gene sequencing, of two unrelated, neurological symptom-free patients who consulted for late-onset of deep multiple cutaneous angiomatoid lesions, are described. RESULTS: The diagnosis of multiple cutaneous angiomatosis was confirmed and related to CCM as detected by MRI in both cases. Analysis of our patients showed normal nucleotide sequences of the genes proposed. CONCLUSIONS: A progressive late-onset of multiple, deep cutaneous venous malformations may indicate the need to investigate a potential coexistence of CCM by MRI. Early diagnosis and prompt treatment is required in these patients. The absence of CCM1, CCM2, and CCM3 mutations might indicate that different genes could be involved in the pathogenesis of these late-onset patients. Careful questioning about family history of CCM is important; our first patient’s daughter had a history of cerebral cavernoma.
Case reports
Cerebral cavernous malformation (CCM), described as abnormally enlarged, thin-walled capillary, raspberry-like cavities mostly affecting the central nervous system, is considered as a life-time risk disorder because of the possible onset of cerebral hemorrhages, seizures, strokes, and focal neurological deficits [1]. Extra-neurological manifestations include retinal and cutaneous vascular malformations [2]. CCM can occur as sporadic (80% of the cases) or familial event (20% of the cases), and the risk of developing neurological symptoms is higher and at a younger age in this latter form [3]. It has been documented a strong correlation between age and number of the cerebral lesions in patients with symptoms and in symptom-free relatives. Familial CCM is consistent with an autosomal-dominant inherited pattern with incomplete clinical penetrance and possible de-novo mutations [1]. This variant is caused by heterogeneous mutations in CCM1 [K-Rev interaction trapped 1 (KRIT1)], CCM2 (MGC4607), and CCM3 (PDCD10) genes, located on chromosomes 7q21-q22 (CCM1), 7p15-p13 (CCM2), and 3q25.2-q27 (CCM3), respectively [4, 5, 6]. According to necroscopy reports or magnetic resonance imaging (MRI) investigations, its frequency reaches 0.5 percent in the general population, but it can exceptionally achieve 50 percent in certain populations such as a Hispano-American series of patients who share a common haplotype linked to a 2105 C→T mutation in the CCM1/KRIT1 gene [1]. We observed two unrelated patients referred to our clinic for the evaluation of multiple asymptomatic cutaneous bluish lesions that appeared in adulthood as violaceous, angiomatoid macules that developed into deep, bluish nodules of varying size.
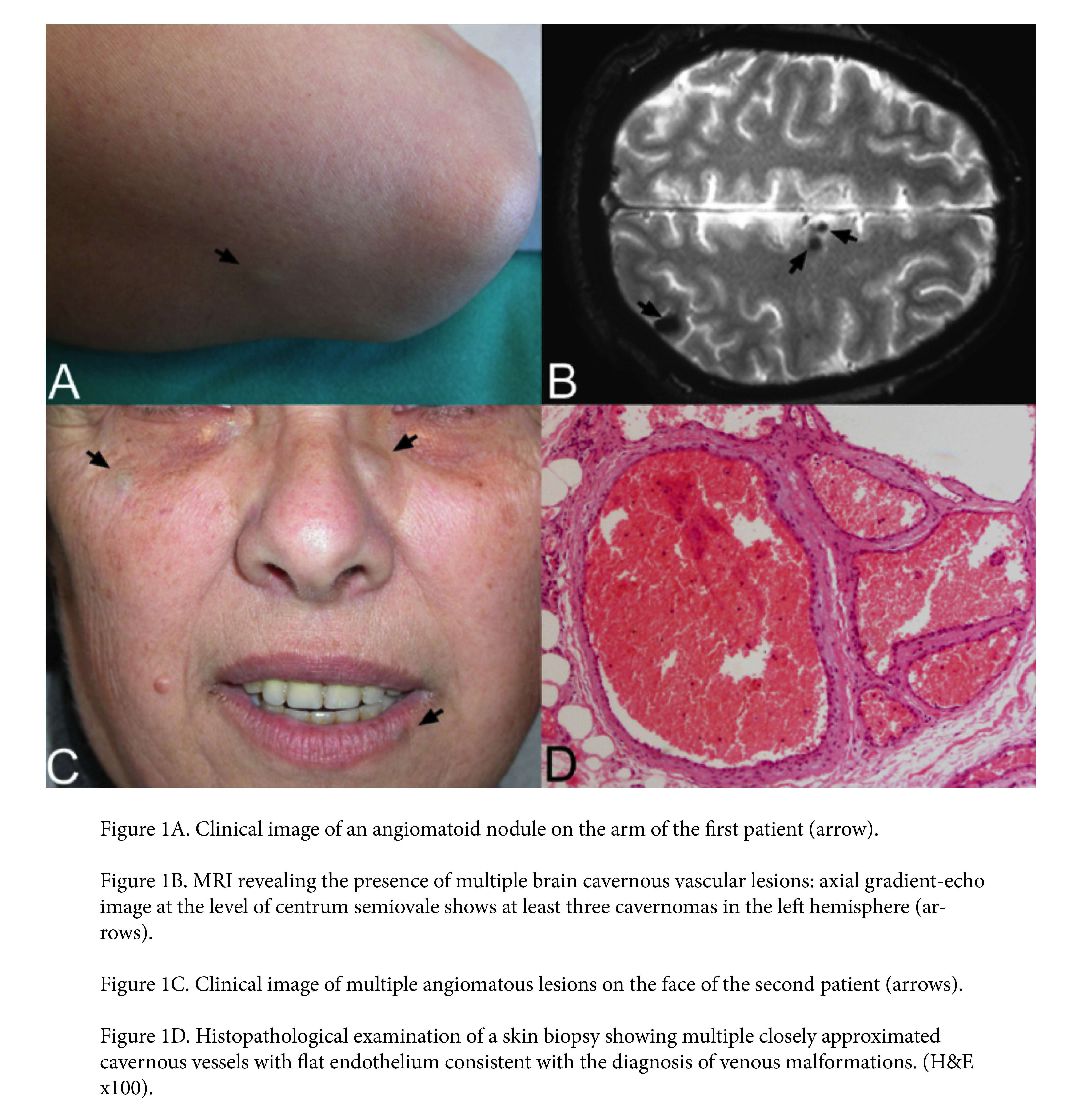 |
| Figure 1 |
|---|
The first patient, a 56-year-old female affected by a not otherwise specified myelitis, had noticed the development of violaceous nodules on her trunk and extremities (Figure 1A) during the last 5 years. Interestingly, her daughter had a history of removal of one cerebral cavernoma. Because of her family history and the onset in her adulthood of angiomatoid lesions gradually increasing in number, we proposed a cerebral MRI, which documented CCM (Figure 1B). The MRI study, performed with a 3 Tesla unit, revealed multiple well-defined lesions in both cerebral and cerebellar hemispheres. Some lesions showed a mixed signal intensity on T1- and T2-weighted images; others were predominantly hypointense especially on gradient-echo images (Figure 1B). The heterogeneous signal intensity or “popcorn-like” appearance is the result of hemorrhages in different stages of evolution, a characteristic finding of CCM. The hypointensity on gradient-echo images reflects susceptibility artifacts consistent with microscopic deposits of hemosiderin and/or calcification, another classical finding of CCM. No other first-degree relatives gave consent to perform similar investigations.
Our second patient, a 43-year-old female, complained of the progressive development of bluish papules over 2 years and frequent episodes of headache. (Figure 1C). The relatively high number, the progressive late-onset in her life, and the size and depth of the vascular lesions suggested the need for a cerebral MRI, which revealed multiple CCM. We proposed similar testing of her first-degree relatives, but their MRIs did not display any anomalies. Similar imaging is planned for her daughter who is now only 2 years old.
Histopathologic examination of two skin lesions confirmed in both patients the diagnosis of multiple cutaneous angiomatosis, revealing the presence, in the deep dermis and subcutis, of large cavernous vessels lined by flat endothelial cells, with little intervening stroma, consistent with venous malformations (Figure 1D).
Discussion
To perform a genetic analysis in our cases, we extracted RNA from angiomatous skin lesions of the two patients, for RT-PCR amplification and direct sequencing. Genomic DNA was obtained from other members of the families to confirm mutations in case of detected genetic abnormalities. We amplified and sequenced KRIT1 coding region (NM_194456), spanning 2211 nucleotides pairs, CCM2 (Malcavernin, NM_001029835; NM_031443), spanning 1335 nucleotide pairs, and CCM3 (Programmed Cell Death 10 PCD10, NM_145859), spanning 693 nucleotides pairs. The analysis has been performed using RNA instead of genomic DNA to easily detect mutations occurring inside intronic sequences, leading to exon skipping or intron insertion mutations. In both patients, the complete analysis of the coding regions of KRIT1 and PCD10 did not show any differences from the wild type sequence.
Our two patients are representative cases of symptom-free CCM. A recent study showed that a two-hit mechanism seems involved in the pathogenesis of CCM that causes a complete loss of the three CCM proteins within the endothelial cells, which line the cavernous capillary cavities [7]. Almost 80 percent of familial CCM patients display a heterozygous germline mutation in one of these three genes [8]. CCM1 behaves as an anti-angiogenic protein necessary to maintain a quiescent human endothelium, thus inhibiting endothelial proliferation, apoptosis, migration, lumen formation, and sprouting angiogenesis in primary human endothelial cells. Because CCM1 strongly induces DLL4-NOTCH signaling, a perturbed NOTCH signaling that is able to induce excessive capillary growth, could induce CCM lesions. Therefore, blocking of NOTCH activity has been proposed as a therapeutic target to ameliorate the CCM1 effects [9]. However, CCM3 silencing may potentially induce enriched capillary-like immature vessel formation in CCM lesions by stimulating endothelial proliferation, migration, and massively growing and branching angiogenic sprouts. Furthermore, CCM2, but not CCM3, silencing has been associated with the different intracranial hemorrhage rate observed from CCM2 and CCM3 mutation carriers [9, 10]. In both patients, the complete analysis of the coding regions of KRIT1 and PCD10, did not show any differences from the wild type sequence. The sequence of the Malcavernin coding region presented high complexity for the presence of different splicing variants. We used primers that amplify the two published isoforms, NM_001029835 and NM_031443, without finding abnormalities. In addition, gel electrophoresis identified, in samples and normal controls, additional bands of lower molecular weight, presumably related to alternative splicing; it was not possible two analyze this in direct sequencing. These alternative splicing variants should not be involved in the pathogenesis of the disease, because the full transcripts were normal and the additional bands were present also in normal unrelated controls. The analysis of our patients showed a normal nucleotide sequence of the genes proposed and analyzed, but other members of the family could not be further investigated. In light of these results, we hypothesize that different genes could be involved, as suggested by the positive family history for CCM; the first patient’s daughter had a history of a cerebral cavernoma. This report underlines the supportive role of the dermatologist in a pre-symptomatic diagnosis of CCM. A progressive late-onset of multiple, often deeply located, cutaneous venous malformations may indicate the opportunity to investigate a potential coexistence of CCM by MRI. If positive, the opportunity presents for early diagnosis and intervention as well as investigation of family members. In light of the histological similarities shared by the cutaneous and cerebral malformations, we wonder if a progressive increase in number and size of the skin lesions, as occurred in our patients, could herald similar behavior in the cerebral localization.
References
1. Labauge P, Laberge S, Brunereau L, Levy C, Tournier-Lasserve E. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Lancet 1998; Dec ;352(9144):1892-1897. [PubMed]2. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol 2009; Sep 23(9):1066-1072. [PubMed]
3. Toll A, Parera E, Giménez-Arnau AM, Pou A, Lloreta J, Limaye N, Vikkula M, Pujol RM. Cutaneous Venous Malformations in Familial Cerebral Cavernomatosis Caused by KRIT1 Gene Mutations. Dermatology 2009;218(4):307-313. [PubMed]
4. Craig HD, Günel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, Ogilvy CS, Berg MJ, Crawford SC, Scott RM, Steichen-Gersdorf E, Sabroe R, Kennedy CT, Mettler G, Beis MJ, Fryer A, Awad IA, Lifton RP. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet 1998 Nov;7(12):1851-8. [PubMed]
5. Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005 Jan;76(1):42-51. [PubMed]
6. Revencu N, Vikkula M: Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet 2006 Sep;43(9):716-21 [PubMed]
7. Clatterbuck RE, Cohen B, Gailloud P, Murphy K, Rigamonti D. Vertebral hemangiomas associated with familial cerebral cavernous malformation: segmental disease expression. Case report. J Neurosurg. 2002 Sep;97(2 Suppl):227-30. [PubMed]
8. Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J. 2010 Mar;277(5):1070-5. [PubMed]
9. Wüstehube J, Bartol A, Liebler SS, Brütsch R, Zhu Y, Felbor U, Sure U, Augustin HG, Fischer A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci U S A. 2010 Jul 13;107(28):12640-5. [PubMed]
10. Schleider E, Stahl S, Wüstehube J, Walter U, Fischer A, Felbor U. Evidence for anti-angiogenic and pro-survival functions of the cerebral cavernous malformation protein. Neurogenetics 2011 Feb;12(1):83-86. [PubMed]
© 2013 Dermatology Online Journal