Striate palmoplantar keratoderma
Published Web Location
https://doi.org/10.5070/D352p3k03xMain Content
Striate palmoplantar keratoderma
Katherine L. White
Dermatology Online Journal 8(2): 16
From New York University Department of DermatologyHistory
This 67-year-old man has had a problem on his palms since early adulthood. The patient is being followed in the Dermatology Clinic at the New York Harbor Health Care System. He reports calluses on his hands, which started in young adulthood; more fingers have become involved with time. His father had corns. He had six siblings without similar findings. None of his four children is affected.
Past medical history includes discoid lupus erythematosus, prostate cancer, infection with hepatitis C virus, a chronic mild macrocytic anemia, and alcohol abuse in the past.
Physical Examination
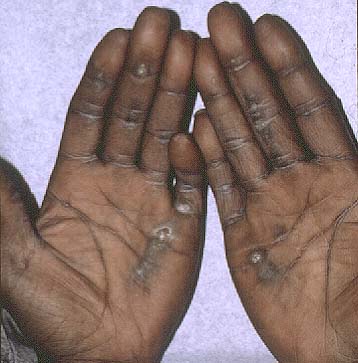 |  |
| Figure 1 | Figure 2 |
|---|
Symmetrically distributed, hyperkeratotic papules were present on the palms, and linear hyperkeratosis extended up the volar aspects of the digits.
Laboratory Data
Hemoglobin was 11.5 g/dl, hematocrit 34.3%, and mean corpuscle volume 96.9 fl. A chemistry profile and liver function tests were normal.
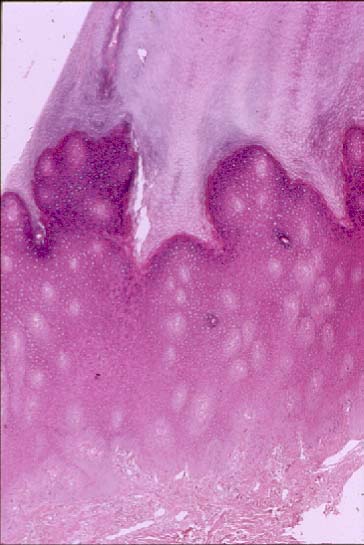
Histopathology
There is marked hyperkeratosis overlying papillated epidermal hyperplasia with hypergranulosis.
Comment
The palmoplantar keratodermas are a heterogeneous group of hereditary disorders of keratinization, which are characterized by epidermal thickening of the palms and soles. Inheritance of the different palmoplantar keratodermas is variable; autosomal dominant, recessive, x-linked, and acquired forms have been identified. Striate keratoderma, also known as Brunaur-Fuhs-Siemens syndrome, is a rare focal palmoplantar keratoderma, with an autotomal dominant pattern of inheritance. Phenotype expression is variable. Onset is usually early in life. Areas of pressure or trauma on the soles are often more affected than are the hands. Hands may show calluses. Manual labor may exacerbate palmar hyperkeratosis.
Recent investigations have identified a gene cluster on human chromosome 18q1 that encodes the desmosomal transmembrane proteins, desmocollins and desmogleins 1-3. These transmembrane proteins are essential components of the desmosome. They belong to the desmosomal cadherin superfamily. Several mutations in the desmoglein 1 gene have been identified as the genetic basis of striatepalmoplantar keratoderma. Mutations in the gene encoding desmoplakin, a desmosomal plaque protein, have also been described in the pathogenesis of striate palmoplantar keratoderma. Thus, striate palmoplantar keratoderma is a genetically heterogeneous disorder of keratinization, which may account for variable clinical manifestations.
Keratolytics are the mainstay of therapy. Use of topical and oral retinoids has also been described.
References
Hennies HC, et al. Localization of a locus for the striated form of palmoplantar keratoderma to chromosome 18q near the desmosomal cadherin gene cluster. Human Mol Genet 4:1015, 1995Helm T, et al. Striate palmoplantar keratoderma: a clinical and ultrastructural study. Cutis 61:18, 1998
Rickman L, et al. Amino-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Human Mol Genet 8:971, 1999
Whittock NV, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol 113:940, 1999
Frank J, et al. Characterization of the desmosomal cadherin gene family: genomic organization of two desmoglein genes on human chromosome 18q12. Exp Dermatol 10:90, 2001
© 2002 Dermatology Online Journal