Giant plexiform neurofibroma in a patient with neurofibromatosis type I
Published Web Location
https://doi.org/10.5070/D350c593b5Main Content
Giant plexiform neurofibroma in a patient with neurofibromatosis type I
Efstathios Rallis MD PhD, Dimitra Ragiadakou MD
Dermatology Online Journal 15 (5): 7
Department of Dermatology, Army General Hospital, Athens, Greece. efrall@otenet.grAbstract
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder primarily affecting the development and growth of nerve cell tissues. Plexiform neurofibroma is considered an uncommon skin tumor. The involvement of the genitourinary tract or the lower limb is rare, with bladder, upper urinary tract and genital involvement reported in decreasing order of frequency. The management of patients with plexiform neurofibroma is not well defined and aiming mostly at controlling symptoms. We present a case of a giant genitourinary plexiform neurofibroma associated with lower limb gigantism in a patient with NF1.
Introduction
Neurofibromatosis types 1 and 2 (NF1, NF2) are autosomal dominant disorders that primarily affect the development and growth of nerve cell tissues [1]. Neurofibromatosis type 1, also known as von Recklinghausen disease, is characterized by various skin lesions and peripheral or central nervous system neoplasms.
Plexiform neurofibroma is considered an uncommon skin tumor [2]; it usually presents at birth or during the first several years of life [3].
We present a rare case of a giant genitourinary plexiform neurofibroma with lower limb gigantism in a patient with NF1.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
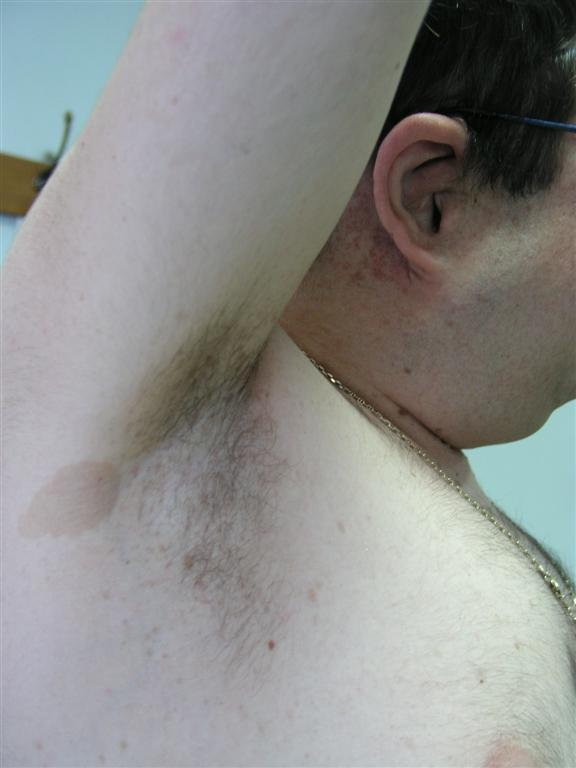
| Figure 1. Gigantic mass involving the right gluteal area and the right lower limb Figure 2. Freckling and café-au-lait patch measuring more than 1.5 cm in diameter in right axilla | |
A 19-year-old male with NF1 was referred to our department in December 2002, to evaluate an enlarging mass involving the right gluteal area and the right lower limb (Fig. 1). The patient also had 11 café-au-lait patches, each measuring more than 1.5 cm in diameter and both axillary (Fig. 2) and inguinal freckling. On the right ear a neurofibroma of approximately 4 cm in diameter was observed (Fig. 3). According to his past medical history, the patient's father and his twin brother also had NF1. The diagnosis of giant plexiform neurofibroma of the genitourinary tract had been made in 1986. During the first five years of his life, the patient had undergone four partial surgical excisions of the tumor with poor results.
 | 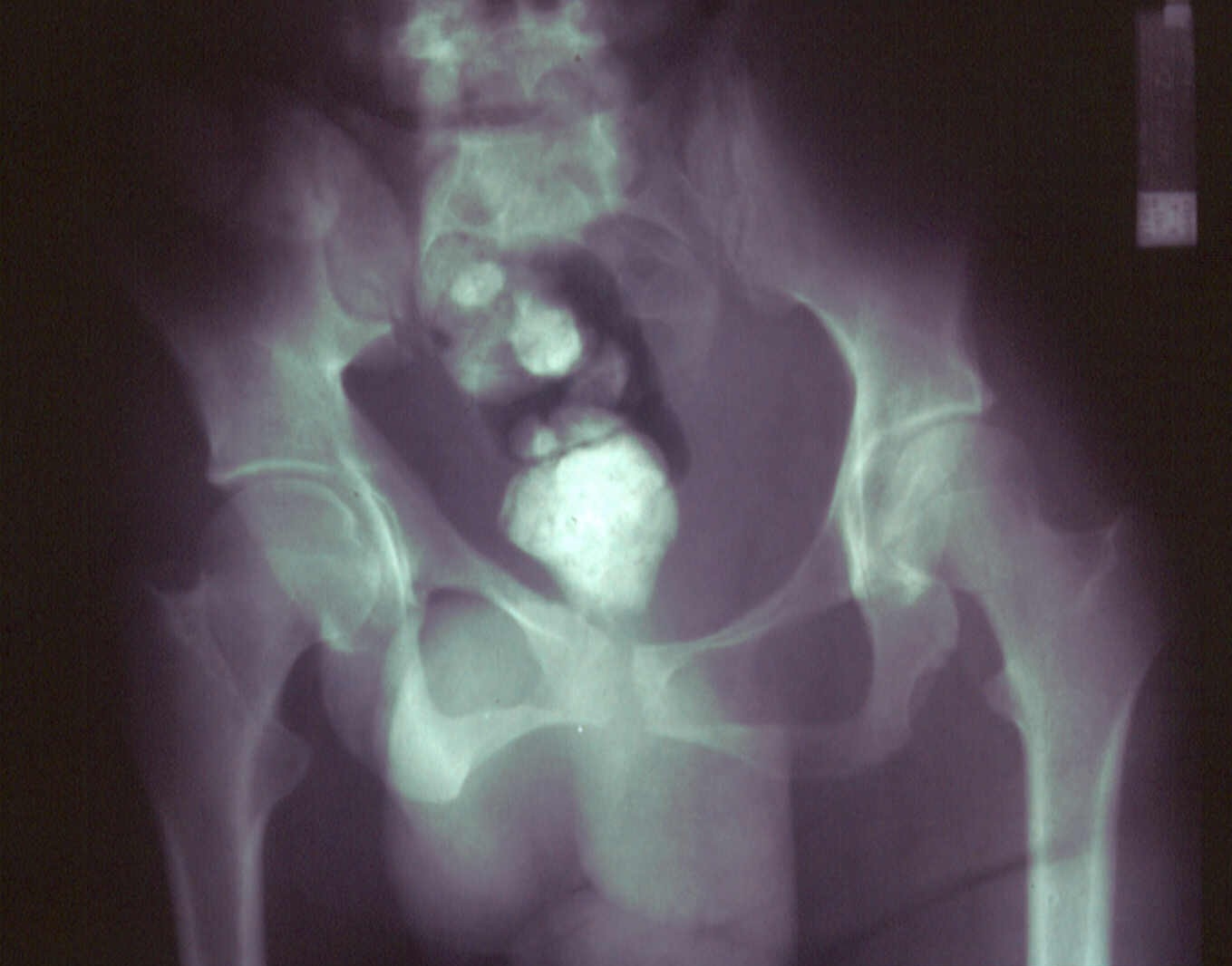 |
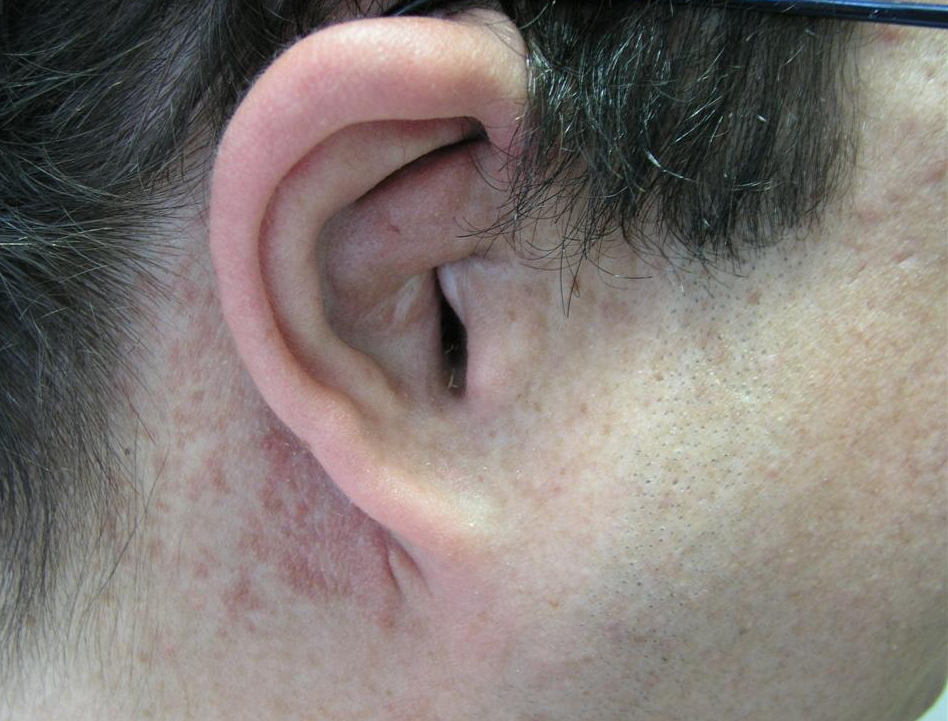
| Figure 3 | Figure 4 |
|---|---|
| Figure 3. Neurofibroma of the right ear Figure 4. (Ro). Enlargement of the right scrotum and thumb-printings in the cortex of the right ischium | |
At the time of his admission in our department, enlargement of the right scrotum (Fig. 4), difficulty with mobility, and disparity in the leg length of the lower extremity were noticed. The physical disfigurement was significantly affecting the patient's quality of life.
A skin biopsy from the right gluteal area was performed and histological examination revealed large, hypertrophic nerves that consisted mainly of spindle-shaped fibroblasts. The nerves were surrounded by a myxoid stroma that extended into the papillary dermis. The findings were compatible with the diagnosis of plexiform neurofibroma with no malignant transformation.
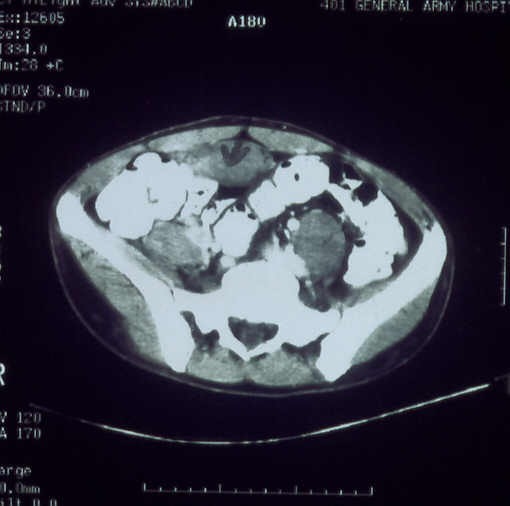 |
| Figure 5 |
|---|
| Figure 5. (CT scan). Extensive mass expanded from the outer space of the bladder to the perineum and the right scrotum |
Blood and urine tests including the vanillylmandelic acid test, were all within normal ranges. Radiological studies showed congenital bowing of the tibia, fibula and lower arm, soft tissue hypertrophy of the right lower extremity, thinning of the right pubis and ischium, enlargement of the right scrotum, and thumb-printings in the cortex of the right ischium due to tumor (Fig. 4). Computer tomography scanning revealed multiple neurofibromas along the roots of lumbosacral plexal and an extensive mass extended from the outer space of the bladder to the perineum and the right scrotum (Fig. 5). Examination of the gastrointestinal tract with gastroscopy and colonoscopy revealed two hyperplastic polyps in the sigmoid colon. Pyelography was unremarkable.
The lower limb gigantism was attributed to the progressive tumor re-growth. Despite the significant physical disfigurement, the patient did not consent to any additional surgical excision.
Discussion
Neurofibromatosis type 1 is an autosomal dominant condition that occurs at an incidence of 1 in 3000 live births [3]. This type accounts for up to 90 percent of the cases of NF [4]. Neurofibromatosis type 2 is a separate autosomal dominant disorder that occurs at an incidence of 1 in 40,000 live births [3].
Neurofibromatosis type 1 has been reported throughout the world. In most large series it has been found that 30-50 percent of NF1 patients have no affected relatives, indicating that the majority of such cases can be attributed to spontaneous mutations [5, 6]. A large number of NF1 mutations have been described; however no genotype-phenotype correlations have been seen except for individuals who have deletions of the entire NF1 gene. Unlike the situation noted in many other dominant disorders, the frequency of new mutations does not appear to increase with advancing paternal age [3].
Among familial cases, it has been suggested that the mode of inheritance of NF1 is autosomal dominant [7]. Although the expressivity of NF1 varies considerably, even among individuals in the same family who are genotypically identical, the penetrance of the disease is essentially 100 percent [3]. The range and severity of clinical findings between relatives with NF1 may be as varied as for those from different families. The specific inherited mutation does not determine the phenotype [7].
The NF1 gene is located on the long arm of chromosome 17q11.2 and encodes a 327 kDa protein called neurofibromin [8]. The exact function of this protein is poorly understood, but the gene encoding neurofibromin has sequence similarities to a group of proteins called GTPase-activating proteins (GAP). This similarity suggests its involvement in negatively regulating the proteins coded by the Ras oncogene [9]. The Ras protein is like other G proteins and is dependent upon GTP binding for its full activity; GAP removes GTP by increasing the conversion of GTP to GDP. Enhanced Ras protein activity has been correlated with human cancer development and dysfunction of neurofibromin could contribute to this [7].
Most NF1 mutations result in reduced intracellular levels of the NF1 gene product, neurofibromin. This appears to be sufficient to cause most of the clinical manifestations of the disease [3].
A consensus development conference was held by the National Institutes of Health (NIH) in 1987 to establish diagnostic criteria of patients with NF1. There were seven diagnostic features recognized at this conference that have been applied without modification during the last 22 years. The diagnosis of NF1 is established when two or more of these seven features listed below are present [10]:
- Six or more café-au-lait macules larger than 5 mm in greatest diameter in prepubertal individuals: 15 mm in greatest diameter in postpubertal individuals.
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- Freckling in the axillary or inguinal regions.
- Optic glioma.
- Two or more Lisch nodules (iris hamartomas).
- A distinctive osseous lesion, such as sphenoid dysplasia or thinning of the long bone cortex, with or without pseudoarthrosis.
- A first-degree relative with NF-1 according to the above criteria.
In our patient, five of the 7 NIH diagnostic criteria were found.
Plexiform neurofibromas are benign peripheral nerve sheath tumors, often involving the trigeminal or upper cervical nerves [11]. They are diffuse, elongated fibromas, histologically similar to discrete neurofibromas and are usually seen in only 5-10 percent of patients with NF1 [7, 12]. They often develop and become physically apparent within the first 2-5 years of life. When present, they are commonly seen on the face and neck [2, 13, 14]. Their growth rate is highly variable. Often, overlying hyperpigmentation ("giant café-au-lait spot") or hypertrichosis can be seen [3].
There are two types of plexiform neurofibromas, nodular and diffuse. Diffuse plexiform neurofibroma is also known as elephantiasis neurofibromatosa and is characterized by an overgrowth of epidermal and subcutaneous tissue associated with a wrinkled and pendulous appearance [4]. Plexiform neurofibromas involving the genitourinary tract or the lower limb are rare, with bladder, upper urinary track and genital involvement reported in decreasing order of frequency [1, 15].
Neurofibroma type 1 is considered a benign tumor condition; however malignant transformation has been reported in 2 percent of patients with NF1 (or 4.2% of those older than 21 years of age) [16]. Malignant peripheral nerve sheath tumors, which generally arise from plexiform neurofibromas, may develop silently in deep plexiform neurofibromas and not give rise to symptoms until distant metastases have occurred. [3].
The management of patients with plexiform neurofibroma is not well defined and is aimed mostly at controlling symptoms. Surgical excision is probably the only therapy available because there is no medication that can prevent or treat plexiform neurofibromas. However the results of surgical excision can be poor and the procedures can be complicated due to the size, location, vascular status, neural involvement, microscopic extension of the tumor, and the high rate of tumor re-growth. Biopsy of plexiform neurofibromas should be performed if the lesions present rapid growth or cause significant pain or focal neurologic dysfunction [3]. In our case it seems that periodic radiological surveillance with CT and psychological support are probably the only reasonable course of action [17].
References
1. Rekha A, Gopalan TR. Von recklinghausen neurofibromatosis-pachydermatocele causing lower limb gigantism: a case report. Int J Low Extrem Wounds 2006; 5: 61-63. [PubMed]2. Khachemoune A, Al Aboud K, Al Hawsawi K. Diffuse plexiform neurofibroma in a 13-year-old girl. Dermatol Online J. 2003; 9(5): 23. [PubMed]
3. Listernick R, Charrow J. The Neurofibromatoses. Wolff K, Goldsmith L, Katz S, Gilchrest B, Paller A, Leffell D. Editors: Fitzpatrick's Dermatology in General Medicine, 7th ed New York: Mc graw Hill ; 2008. p.1331-1339.
4. Harper JI. Genetics and genodermatoses. In: Champion RH, Burton JL, Burns DA, Breathnach SM editors Rook/Wilkinson/Ebling. Text book of Dermatology. 6th ed. Oxford: Blackwell Science; 1998; 378-384.
5. Samuelsson B, Axelsson R. Neurofibromatosis. A clinical and genetic study of 96 cases in Gothenburg, Sweden. Acta Dermatol Venereol Suppl 1981; 95: 67-71. [PubMed]
6. Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Rechklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 1989; 26: 704-711. [PubMed]
7. Tsao H. Neurofibromatosis and tuberous sclerosis. In: Bolognia JL, Jorizzo JL, Rapini RP. Editors: Dermatology. Vol I. Edinburgh: Mosby, 2003: p 853-867.
8. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet 1996; 33: 2-17. [PubMed]
9. Wigler MH. Oncoproteins. GAPs in understanding Ras. Nature 1990; 346: 696-697. [PubMed]
10. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988; 45(5): 575-578. [PubMed]
11. Korf BR. Plexiform neurofibromas. Am J Med Genet 1999; 89: 31-37. [PubMed]
12. Pivnick EK, Riccardi VM. The Neurofibromatoses. In : Freedberg IM, Eisen AZ,Wolff K, Austen KF, Goldsmith LA, Kartz SI, Fitzpatrick TB. Fitzpatrick's Dermatology in General Medicine. New York: Mc graw Hill ; 1999; 2152-2158.
13. Fisher DA, Chu P, McCalmont T. Solitary plexiform neurofibroma is not pathognomonic of von Recklinghausen's neurofibromatosis: a report of a case. Int J Dermatol 1997; 36: 439-442. [PubMed]
14. Gutmann DH, Collins FS. Neurofibromatosis type 1. In: Vogelstein B, Kinzler KW, eds. The genetic basis of human cancer. New York, McGraw-Hill, 1998: 423-442.
15. Kaefer M, Adams MC, Rink RC, Keating MA. Principles in management of complex pediatric genitourinary plexiform neurofibroma. Urology 1997; 49: 936-940. [PubMed]
16. Voutsinas S, Wynne-Davies R. The infrequency of malignant disease in diaphyseal aclasis and neurofibromatosis. J Med Genet 1983; 20: 345-349. [PubMed]
17. Nahabedian MY, Rozen SM, Namnoum JD, Vander Kolk CA. Giant plexiform neurofibroma of the back. Ann Plast Surg 2000; 45: 442-445. [PubMed]
© 2009 Dermatology Online Journal