Long-term AL primary amyloidosis. A case report.
Published Web Location
https://doi.org/10.5070/D34w95m3xnMain Content
(6) Long-term AL primary amyloidosis. A case report
by
Dermatology Online Journal, December 1995
Volume 1, Number 2
ABSTRACT
This is a case report of AL primary amyloidosis with involvement limited to the skin for more than twenty years before the development of internal organ involvement. The clinical features suggested the systemic form of amyloidosis, rather than that found with nodular cutaneous amyloidosis.
Introduction
Longstanding lesions of nodular amyloidosis even in the face of a negative workup for internal involvement, can be consistent with systemic disease. This is an example of a patient who had extensive cutaneous amyloidosis for twenty years before developing cardiac involvement which then lead to her death.Patients who present with cutaneous features of amyloidosis usually undergo an evaluation to categorize the disease. Skin biopsy with ultrastructural immunohistochemical stains can help confirm the presence and subtype of amyloid. Once a patient has been found to have cutaneous amyloidosis, an extensive workup must be done to evaluate patients for the possibility of systemic involvement. Workup for systemic disease includes: serum and urine immunoelectrophoresis, rectal mucosal or abdominal fat biopsy, bone marrow biopsy, CBC, chemistry panel, and skull and spine X-rays. The origin of the amyloid and the extent of internal involvement are used to determine the approach to treatment.
CASE REPORT
A seventy-two-year-old Caucasian female complained of easy eyelid bruising for over twenty years. Her past medical history was remarkable for bilateral carpal tunnel syndrome treated surgically several years prior to presentation to the clinic. Her other medical problems were hypothyroidism and hypertension. The patient's medications included Synthroid, Procardia and Ecotrin, which she had been taking for two years. An eyelid skin biopsy revealed amyloid deposit. Laboratory evaluation for systemic involvement included normal serum chemistry and electrocardiogram, CBC, skull x-ray and total X-ray series. Biopsies of rectal mucosa and polyps from her colon did not reveal any evidence of amyloid. An echocardiogram, performed 4 years earlier,was normal.
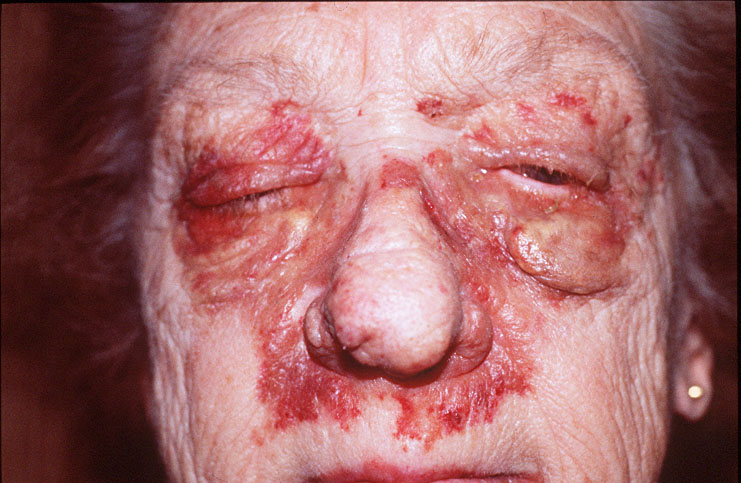
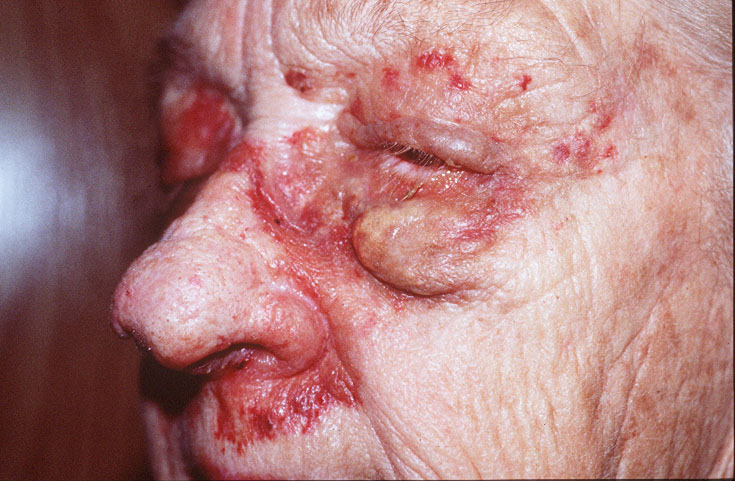
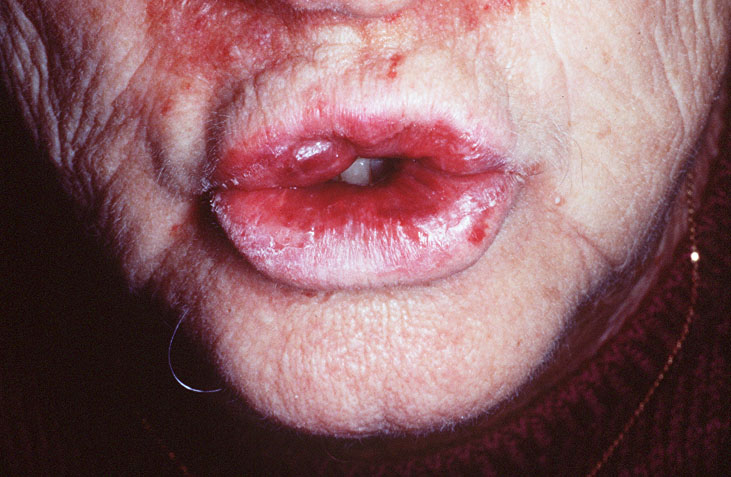
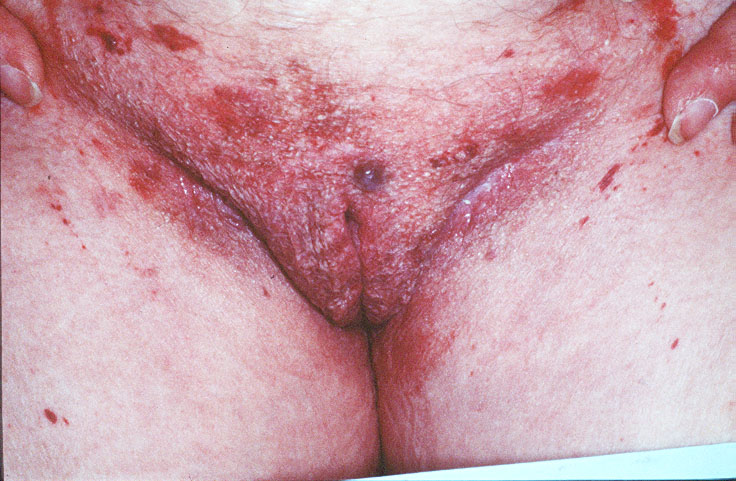
Cutaneous examination revealed tumefactive ecchymotic papules and plaques involving the periorbital, central face, lips, inframammary and groin regions. Her tongue was not enlarged.
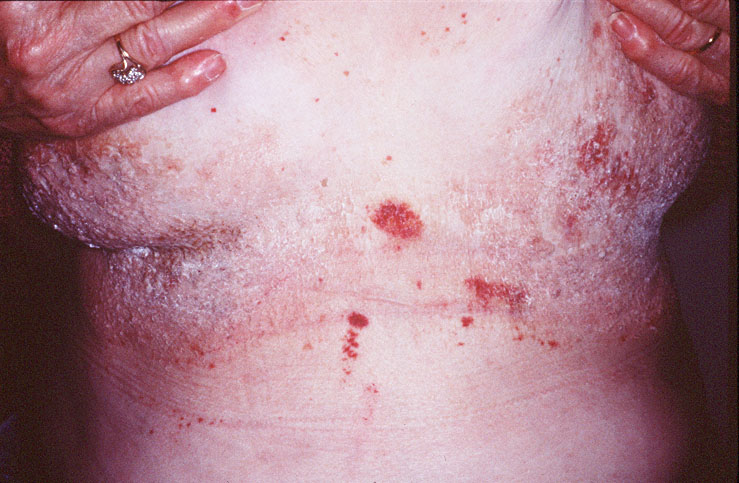
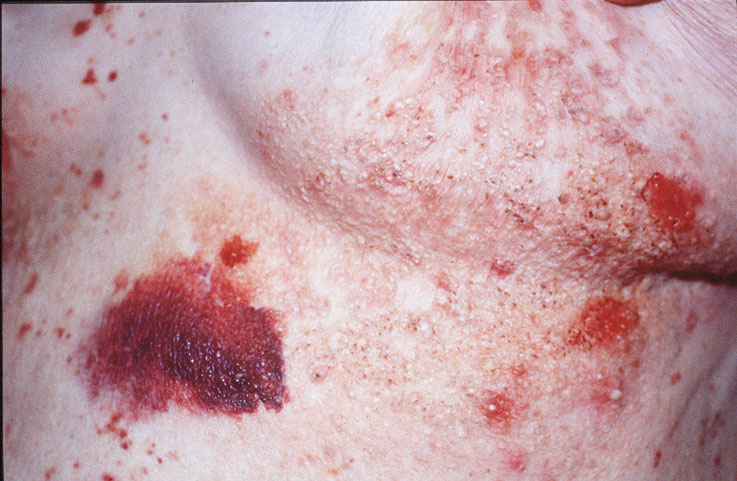
Skin biopsy of a typical lesion on the patient's face showed a pale eosinophilic material distributed throughout the dermis. Congo red staining with polarization revealed apple green birefringence characteristic of amyloid. Thioflavin-T showed brilliant orange fluorescence, also characteristic of amyloid. Electron microscopy showed six to ten nanometer non-branching fibrils diagnostic of amyloid.
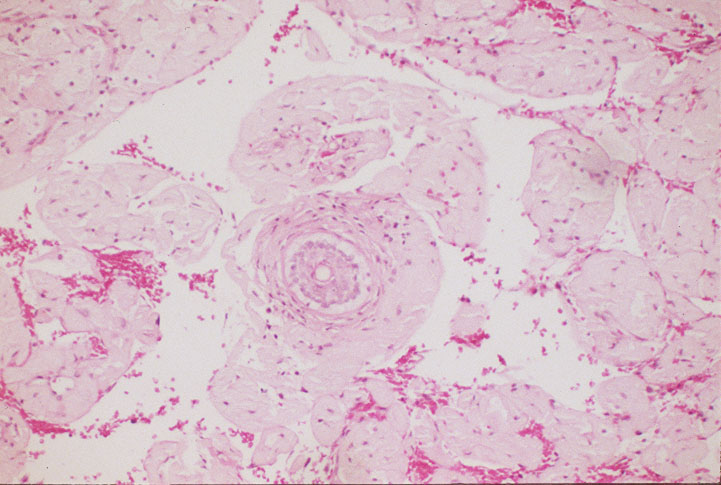
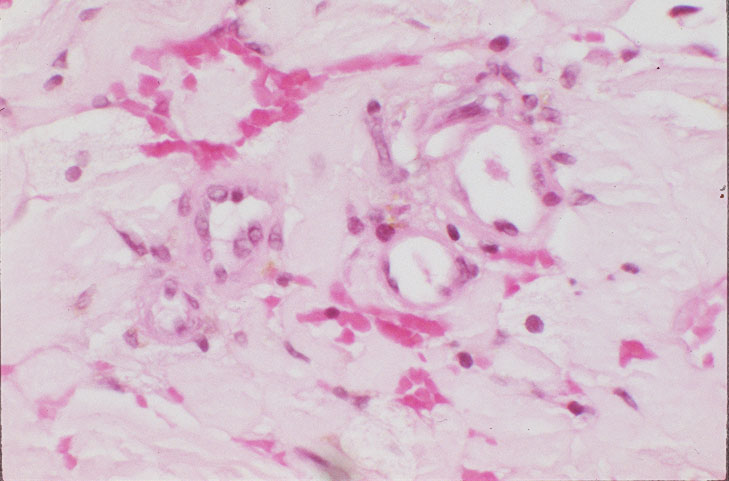
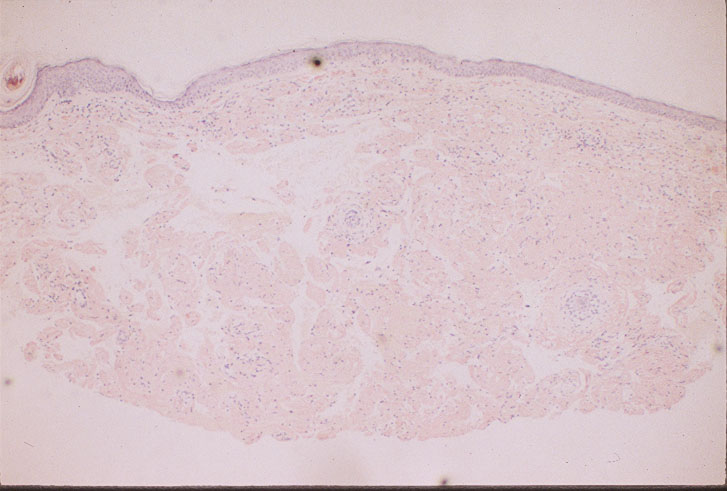
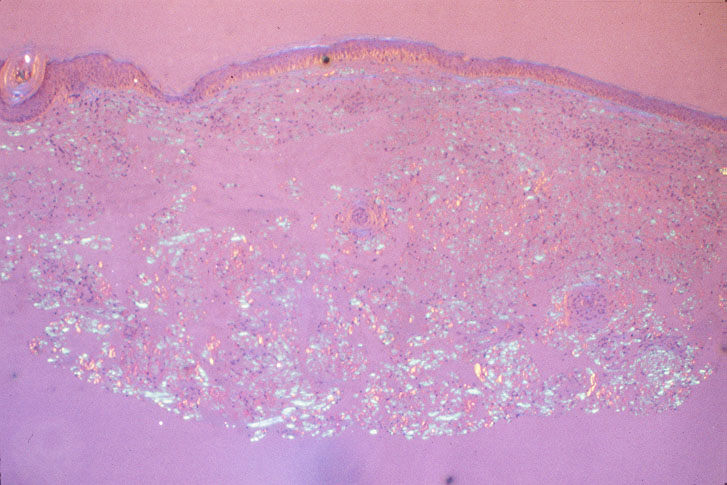
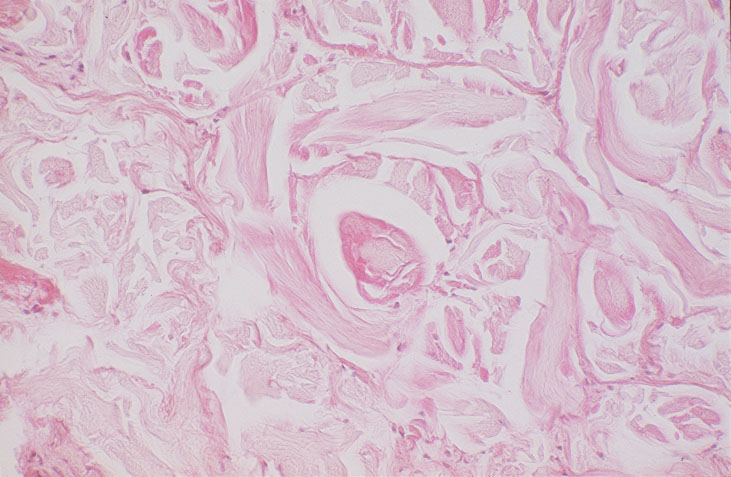
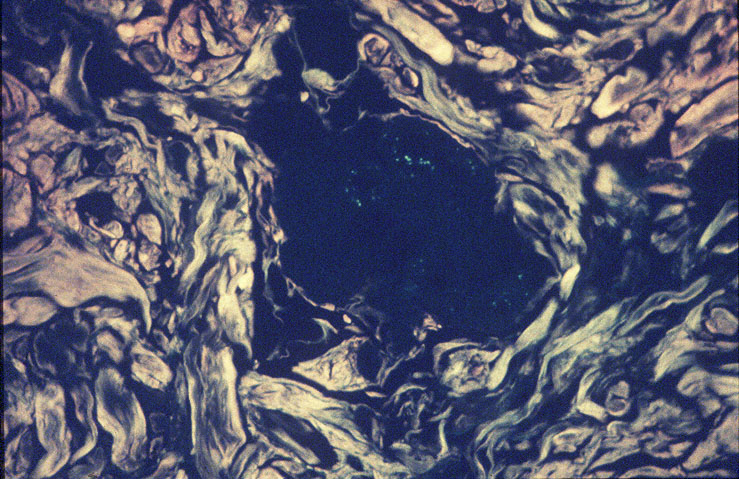
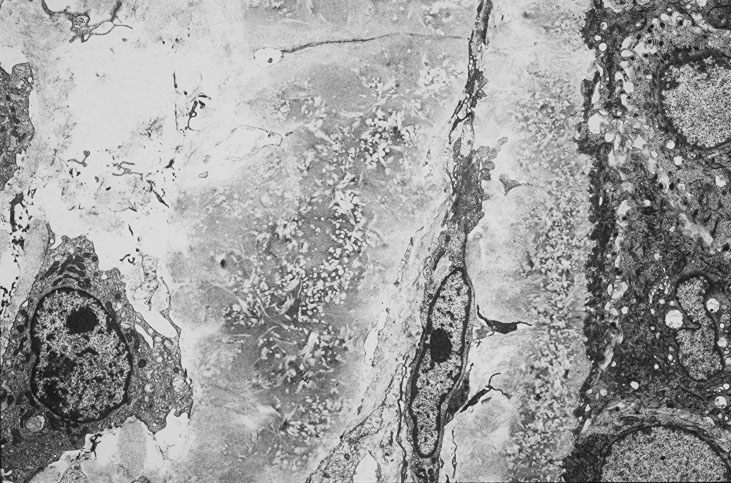
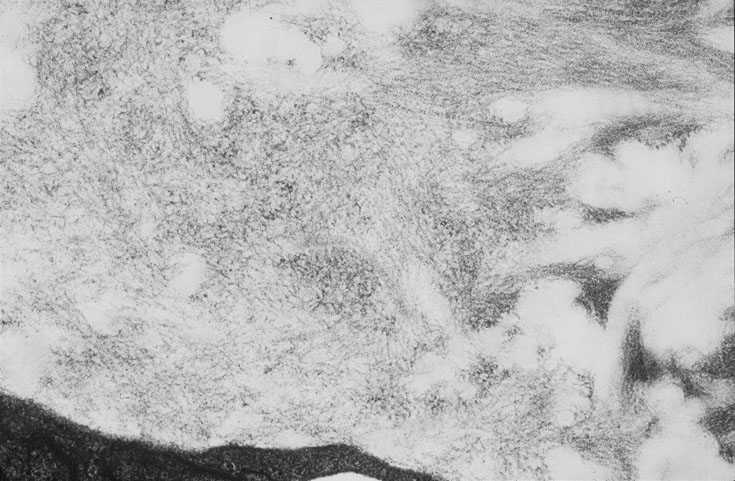
A second work-up was initiated for systemic disease. Serum electrophoresis now revealed a lambda-light chain spike. Immunoglobulin electrophroesis revealed somewhat low levels of IgG A & M, but no evidence of paraprotein. Twentyfour-hour urine protein was normal. Bone marrow biopsy and cytogenetics were normal. Immunohistochemical stains were negative for the cytokeratin kappa light chain, but there was diffuse staining for lambda light chain throughout the dermis. The echocardiogram now revealed left ventricular hypertrophy and mitral regurgitation consistent with amyloid infiltration.
The evaluation suggested AL type of amyloidosis and the clinical history and previous work-up indicated that for twenty years her disease had been confined to the skin, but now had cardiac involvement. The most consistent diagnostic labels for her condition varied between nodular cutaneous amyloidosis (without nodules) and primary systemic amyloidosis (without systemic involvement for 20 years). After following the patient for an additional 2 years, she developed symptoms of congestive heart failure and died from cardiac failure. Subsequent cardiac muscle biopsy revealed amyloid infiltration.
DISCUSSION
The clinical pattern of amyloid involvement with the skin of this patient was consistent with systemic amyloidosis, however her history of long-term involvement was unusual. Because of the initially negative systemic work-up, the lambda light chains deposited throughout her dermis, and her long survival time, we considered a diagnosis of primary cutaneous amyloidosis.This patient initially presented with amyloidosis which was confined to the skin. There are three types of primary cutaneous amyloidosis: nodular cutaneous amyloidosis, macular amyloidosis and lichenoid amyloidosis. These conditions can be distinguished on both clinical and histologic grounds. Macular and lichenoid forms of primary cutaneous amyloidosis occur relatively commonly and are associated with pruritus and friction. They generally consist of lichenoid papules and plaques located on the extensor surfaces. Histologically, the macular and lichenoid forms of this disease have amyloid deposits confined to the papillary dermis. Immunohistochemistry stains are positive for cytokeratin. Nodular cutaneous amyloidosis often presents with facial involvement. Biopsies demonstrate amyloid deposits throughout the dermis and immunohistochemistry is negative for cytokeratin and positive for lambda and or kappa light chains. Thus, of these three conditions and with the presence of lambda-light chains in her dermis, her findings were most consistent with nodular cutaneous amyloidosis (NCA).
NCA is a rare condition with fewer than fifty reported cases in the World's literature. It usually occurs in the sixth and seventh decades of life. Lesions typically involve the face, scalp, and intertriginous regions. Lesions usually consist of one or two isolated tumefactive, purpuric plaques which can be waxy appearing and variable in color and size. NCA progression to systemic disease has been estimated to occur in fifteen to fifty percent of patients.
The problem with using a diagnosis of NCA for this patient was that her lesions did not consist of the typical isolated tumefactive nodules and plaques. The pattern of her lesions were more consistent with systemic amyloidosis, and since she ultimately developed systemic involvement, a diagnosis of systemic amyloidosis was entertained.
Systemic amyloidosis is a rare disorder which usually occurs in aged persons and has a poor prognosis. Systemic amyloidosis can be primary, associated with multiple myeloma, or secondary to another disease. The overall median survival time for 19 patients with systemic amyloidosis was reported to be only eight months.(1) Analysis of the amyloid deposits allow for classification of the systemic amyloidosis. The different types of amyloid proteins deposited are related to apolipoproteins (AA type amyloidosis), immunoglobulin light chains (AL amyloidosis), beta 2-microglobulin, and transthyretin.
The secondary form of systemic amyloidosis is usually associated with AA type deposition and may be due to a variety of illnesses including rheumatic disease, infectious disease, and inflammatory bowel disease. Patients with secondary amyloidosis tend to present with renal, gastrointestinal or cardiac complications, and have a median survival time of 24.5 months.(2)
Primary systemic amyloidosis is usually associated with the deposition of immunoglobulin light chains (AL), without an underlying plasma-cell dyscrasia. The reported median survival time for the 859 primary systemic amyloidosis patients at the Mayo Clinic was 2.1 years.(3) There is one report of one patient with 18 year survival.(4)
The patient presented here was eventually diagnosed with AL type primary systemic amyloidosis. Because she had no evidence of cardiac, gastrointestinal, or renal involvement for 20 years, her clinical diagnosis was initially thought to be a variant of nodular cutaneous amyloidosis. However, the clinical presentation of skin involvement was most consistent with the systemic form rather than NCA. We think a better term for her initial presentation would be 'AL primary cutaneous amyloidosis.'
REFERENCES
(1) Wong CK, Wang WJ. Systemic amyloidosis. A report of 19 cases. Dermatology, 1994, 189(1):47-51.(2) Gertz MA, Kyle RA. Secondary systemic amyloidosis: response and survival in 64 patients. Medicine, 1991 Jul, 70(4):246-56.
(3) Gertz MA, Kyle RA. Amyloidosis: prognosis and treatment. Seminars in Arthritis and Rheumatism, 1994, Oct, 24(2): 124-38.
(4) Wallis MS, Stough DB. Extended survival of patients with primary systemic amyloidosis. Cutis, 1992 Mar, 49(3):193-5.
All contents copyright (C), 1995. Dermatology Online Journal University of California Davis