Leopard Syndrome
Published Web Location
https://doi.org/10.5070/D34p76479rMain Content
Leopard Syndrome
R Porciello MD1, L Divona MD1, S Strano MD2, A Carbone MD1, C Calvieri MD1, S Giustini MD1
Dermatology Online Journal 14 (3): 7
1. University of Rome "La Sapienza," Department of Dermatology, Policlinico "Umberto I," Viale del Policlinico, 155 Rome,
Italy. annacarb1979@libero.it 2. University of Rome "La Sapienza," Department of Cardiovascular and Respiratory FisiopatologyAbstract
The L.E.O.P.A.R.D. syndrome is an autosomal, dominant disorder with characteristic features that include: multiple lentigines, café au lait spots, electrocardiographic conduction abnormalities, ocular hypertelorism, obstructive cardiomyopathy, pulmonary stenosis, abnormal (male) genitalia, retardation of growth, and deafness. Patients do not usually present all the clinical features traditionally associated with the disorder. Indeed, several features are not present until late in life and do not become clinically manifest until puberty. It has been observed that this syndrome is caused by a "missense" mutation in PTPN11, a gene encoding the protein tyrosine phosphatase SHP-2 located on chromosome 12q22. A diagnosis of LEOPARD syndrome may be established exclusively on the basis of clinical criteria. In our case, the patient was diagnosed with the syndrome late in his life when he was already exhibiting all its distinctive clinical features. We have reported the case of a LEOPARD syndrome patient exhibiting extremely elongated vertebral and basilar arteries previously undescribed in the literature.
Introduction
The L.E.O.P.A.R.D. syndrome is an autosomal, dominant disorder with characteristic features that include: multiple lentigines, café au lait spots, electrocardiographic conduction abnormalities, ocular hypertelorism, obstructive cardiomyopathy, pulmonary stenosis, abnormal (male) genitalia, retardation of growth, and deafness.
This disorder is also known as cardiomyopathic lentiginosis, multiple lentigines syndrome, cardio-cutaneous syndrome or Moynahan syndrome. However the name LEOPARD syndrome has now been firmly established in the clinical literature.
The LEOPARD syndrome was first described by Zeisle and Becker in 1935. Similar cases were later reported by Moynahan and Walther, but it was Gorlin, in 1969, who first introduced the name "LEOPARD syndrome" [1].
Patients do not usually present all the clinical features traditionally associated with the disorder. Indeed, several features are not present until late in life and do not become clinically manifest until puberty. In 1984, Colomb and Morel reported the following findings in 38 LEOPARD syndrome patients: lentiginosis (100%), electrocardiographic abnormalities (80%), skeletal abnormalities (60%), hypertelorism (50%), short stature (42%), mental retardation (35%), abnormalities of male genitalia (29%), and deafness (27%).
The most frequently reported heart defect is pulmonary stenosis which is observed in 40 percent of the cases (Gorlin, 1990) [2].
The LEOPARD syndrome is characterised by highly variable phenotypic expressivity and is diagnosed in the presence of multiple lentigines and at least two other characteristic features. If lentigines are absent, at least three other clinical features must be present (Voron et al. 1976) [3]. In such cases, especially in small children, careful differentiation from the Noonan syndrome is needed as both disorders are characterised by heart defects, retardation of growth and facial dysmorphism [4]. It has been observed that both syndromes are caused by a "missense" mutation in PTPN11, a gene encoding the protein tyrosine phosphatase SHP-2 located on chromosome 12q22 [5]. This protein plays an important role in the transduction of intracellular signals for several growth factors, cytokines and hormones [6]. The PTPN11 mutations most frequently reported in LEOPARD syndrome patients are located in exon 7 (Tyr279Cys) and exon 12 (Thr468Met). Recently, a new mutation (Gln506Pro) has been detected in exon 13 [7]. However, the mutations most frequently recurring in the Noonan syndrome [8] (50% of all the cases) are found in exon 3 and exon 8 [9].
The etiopathogenesis of this disorder remains unknown. According to some authors, LEOPARD syndrome patients have increased melanocytic activity secondary to an abnormal development of neural crest cells and increased beta-adrenergic effector activity in the myocardium [10].
Histological examinations of skin lesions revealed melanocytic hyperplasia and a related increase in melanin production. Electron microscopic examination revealed accumulations of melanosomes (autophagosomes) in melanocytes; they contain giant granules of pigment similar to those observed in the café au lait spots associated with neurofibromatosis [11].
Case Report
A 65-year-old man was referred to us with erythematous desquamative lesions in the crural region. He underwent direct microscopic and culture examinations testing positive for candida. The physical examination of the patient's skin revealed generalized lentiginosis (Fig. 1) that did not affect either the oral mucosa or the genitalia. It also revealed ocular hypertelorism, slight prognathism (Fig. 2), webbed neck, pectus carenatum, short stature, deaf-mutism and hypospadia.
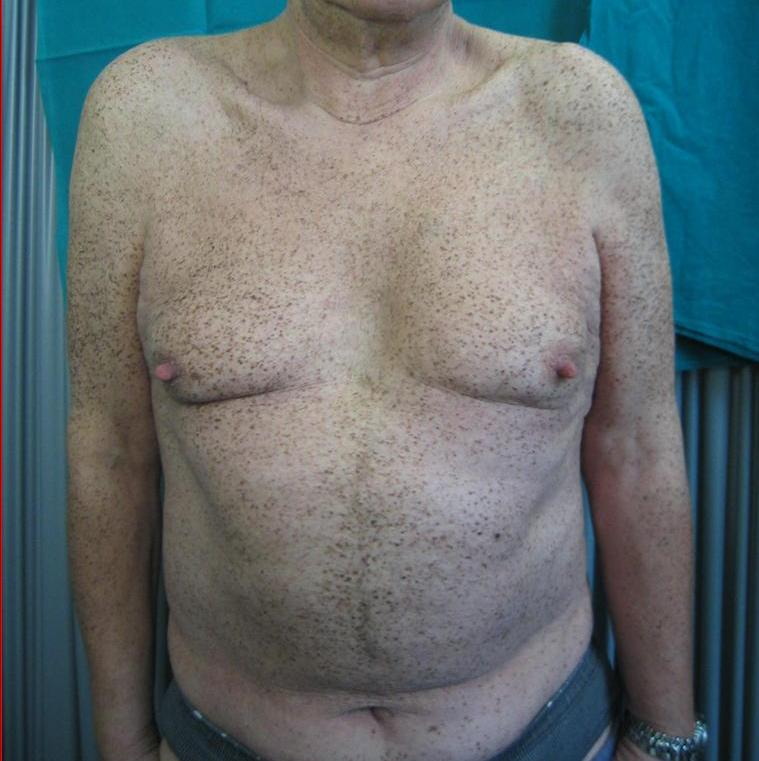 | 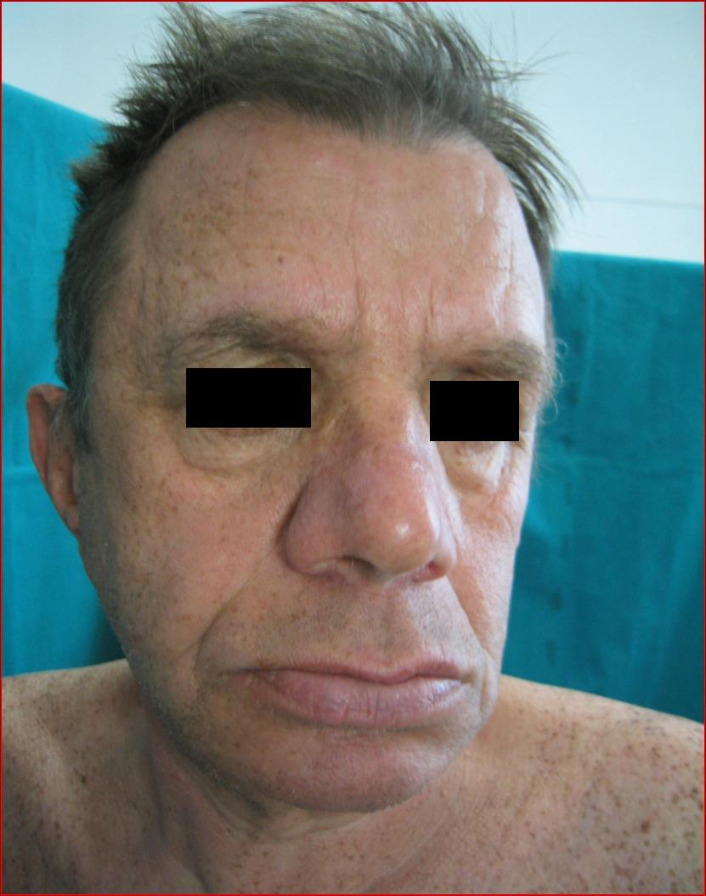 |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. Generalized lentiginosis Figure 2. Facies | |
The peculiarity of the clinical findings prompted the performance of a series of diagnostic and laboratory tests. Tests and examinations included:
- Otolaryngological consult: opaque, severely retracted tympanic membranes with atelectasic areas, especially in the right ear; nasal septum deviated to the right with hypertrophic turbinates; consequences of previous uvulo-palato-plasty and tonsillectomy.
- Audiovestibular testing: profound sensorineural hearing loss in the left ear and anacusis in the right ear.
- Impedance testing: type C tympanogram; absence of evoked stapedial reflexes.
- Dynamic ECG (Holter monitor): moderate sinus tachycardia throughout the recording period; normal physiological heart rate decrease during night sleep; sporadic atrial ectopic beats; sporadic isolated ventricular ectopic beats; stable left intraventricual conduction delay.
- Echocardiogram: left ventricle of normal size and thickness; normal and uniform global kinesis; aortic root of normal size; normal valvular opening; enlarged left atrium; mitral valve (mitral valve cusps close evenly); right ventricle of normal size and function; right atrium of normal size; mild mitral and tricuspid valve regurgitation.
- Chest CT showing: a solid, hypodense nodular formation of probable fibrous nature with a sharp outline and about 1.5 cm in maximum diameter without a considerable increase in densitometry values after contrast medium administration and adjacent to the initial tract of the descending thoracic aorta at the level of the lower left lobe apex; at the level of the abdomen, liver size at the upper limits of normal without focal alterations of densitometry levels; at the level of the right kidney upper pole, a nodular formation of heterogeneous density, about 9 cm in diameter which may be associated with an angiomyolipoma.
The patient also reported a persistent headache and, as a result, underwent:
- Cranial CT Angiography: extremely elongated left vertebral artery and basilar artery with a tortuous pattern at the level of the tip; moderate (especially left) ventricular dilatation.
- Magnetic resonance imaging (MRI) of encephalon and MRI angiography: cortical-subcortical, supratentorial, bi-hemispherical atrophy associated with a meningitis characterised by a chronic development of the cranial vault and base; rarefaction of the supratentorial white matter; elongated basilar artery with no evidence of endoluminal thrombotic vegetations nor aneurismal dilatations; neither areas of intraparenchymatous lesions nor consequences of a previous blood effusion are observed.
Mutation screening for the PTPN11 gene was performed in our patient: we extracted DNA from the peripheral blood lymphocytes and we screened the entire PTPN11 gene coding region for mutatios. All fragments were amplified by polymerase chain reaction (PCR). Denaturing high-performance liquid chromatography (DHPLC) screening of PCR products was done using the WAVE DNA Fragment Analysis System (Transgenomic). Bidirectional direct sequencing of the purified PCR product of the conspicuous fragment with exon 7 was performed using the ABI Big Dye Terminator Sequencing kit and an ABI PRISM 377. The formula of the mutation, already known to be associated with the phenotype of LEOPARD syndrome, is as follows: 836A-G in codon 279 (Tyr279Cys). These findings confirm the diagnosis of LEOPARD syndrome.
Discussion and Conclusion
A diagnosis of LEOPARD syndrome may be established exclusively on the basis of clinical criteria. In our case, the patient was diagnosed with the syndrome late in his life when he was already exhibiting all its distinctive clinical features.
It should be pointed out that lentiginosis is the most frequently occurring feature, being observed in 100 percent of LEOPARD syndrome patients, followed by electrocardiographic abnormalities (80%), skeletal abnormalities (60%), hypertelorism (50%), short stature (42%), mental retardation (35%), abnormal male genitalia (29%), and deafness (27%).
The most frequently observed heart defect is pulmonary stenosis, described in 40 percent of the cases (Gorlin, 1990).
Clinically, lentigines are dark brown in color, irregularly shaped, and can range in size from that of a lentil, to 5 mm in diameter. They are usually found on sun-exposed areas and periorificial areas; they are less frequently observed on palms, soles and genitalia. The oral mucosa is usually unaffected. Distinct facial features are usually exhibited by LEOPARD syndrome patients: triangular shaped face due to frontal swellings, hypertelorism and low-set ears. Most patients have lower-than-average weight and stature. Some skeletal abnormalities may be observed, including pectus excavatum (sunken chest) or carinatum (protruding breastbone), scoliosis, absence of ribs, abnormal elbow articulation. Mild mental retardation is observed in some cases, although intellectual faculties may be normal in LEOPARD syndrome patients. Cardiac abnormalities include electrocardiographic conduction defects and anatomical malformations; electrocardiographic conduction abnormalities are especially frequent. Axial deviation, prolonged PR interval, left anterior hemiblock (LAH), bundle-branch block and complete heart block (CHB) are also described. Hypertrophic cardiomyopathy seems to be the most frequently reported anatomical anomaly [12]: the most frequently described valvular lesion is subaortic stenosis. About 50 percent of male patients exhibit hypospadias. Unilateral cryptorchidism may also be present. Female patients may have missing ovaries or unilateral ovarian hypoplasia.
The presence of multiple lentigines should not be ignored; these may be evidence of other characteristic syndromes, including the Cronkhite-Canada syndrome, the Carney complex, and the Bandler syndrome, in which lentiginosis is circumscribed. The LEOPARD syndrome, like the Carney complex (LAMB and NAME syndromes, myxomas, lentigines, endocrine disorders, melanocytic schwannoma), another autosomal, dominant disorder, is characterised by generalized lentiginosis associated with other systemic anomalies. Clinically, the lentigines observed in the Carney complex are similar to those described in the LEOPARD syndrome and are present all over the patient's body: the only difference is that lentigines also appear on the oral mucosae unlike in the LEOPARD syndrome. On the contrary, facial dysmorphism, which is a characteristic feature of the LEOPARD syndrome, is absent in the Carney complex [13].
The clinical evaluation of the LEOPARD syndrome is based on an ECG, a chest X-ray and an echocardiogram. The prognosis is mainly determined by the nature and severity of the cardiopulmonary lesions. Patients should undergo periodic ECG, chest X-ray and echocardiographic examinations, depending on their clinical picture.
When Gorlin et al. first introduced the name "LEOPARD" some thirty years ago, the authors hoped that "careful family studies would clarify the minor manifestations of this syndrome," although they already suspected "it will not be possible to define the limits of the syndrome." Today, new "clinical abnormalities" (compared to those previously defined by Voron et al.) are still being found in LEOPARD syndrome patients, confirming the highly variable expressivity and penetrance of the disease. This may be the result of a mutation in PTPN11, a gene encoding the protein tyrosine phosphatase SHP-2: this protein plays a key role in intracellular signal transduction pathways and interacts with the angiopoietin-1 receptor, essential for both angiogenesis and the signaling cascade of the endothelial growth factor [14].
Moreover, angiopoietin-1 has a mitogenic effect on endothelial smooth muscle cells induced by nitric oxide (NO) [15]. Such interactions may account for the vascular manifestations observed in our patient.
We have reported the case of a LEOPARD syndrome patient exhibiting extremely elongated vertebral and basilar arteries previously undescribed in the literature. This finding may be explained by providing a better description of the functions of protein tyrosine phosphatase SHP-2, encoded by the PTPN11 gene (involved in the development of the disorder), so as to establish a better genotype-phenotype correlation. The PTPN11 mutation itself may account for the highly variable expressivity and penetrance of the syndrome.
The patient was diagnosed with the syndrome rather late in life by dermatologists, although he had been previously evaluated by other specialists who failed to associate all of the findings with the LEOPARD syndrome. The authors emphasize that this case is exceptional insofar as life expectancy was longer than other LEOPARD syndrome cases described in previous reports; these have had an early mortality due to cardiopathies [16].
References
1. Gorlin RJ, Anderson RC, Blaw M. 1969. Multiple lentigines syndrome. Am J Dis Child 117:652-662. PubMed2. Chong WS, Klanwarin W, Giam YC. Generalized Lentiginosis in Two Children Lacking Systemic Associations: Case Report and Review of the Literature. Pediatric Dermatology Vol. 21, N° 2, 139-145, 2004. PubMed
3. Voron DA, Hatfield HH, Kakhoff RK. 1976. Multiple lentigines syndrome. Case report and review of the literature. Am J Med 60;447-456. PubMed
4. Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein Kinase signal transduction. Human Molecular Genetics, 2006; Vol 15, N° 2; 220-226. PubMed
5. Hanna N, Montagener A, Lee WH, Miteva M, Vidal M, Vidaud M, Parfait B, Raynal P. Reduced phosphatase activity of SHP-2 in LEOPARD Syndrome: Consequences for PI3K binding on Gab1. FEBS Letters 580 (2006); 2477-2482. PubMed
6. Legius E, Schrander-Stumpel C, Shollen E, et all. PTPN11 mutations in LEOPARD sindrome. J Med Genet 2002; 39; 571-574. PubMed
7. Conti E, Dottorini T, Sarkozy A, Tiller GE, Esposito G, Pizzuti A, Dallapiccola B. A Novel PTPN11 Mutation in LEOPARD Sindrome. Human Mutation 2003. PubMed
8. Digilio MC, Sarkozy A, De Zorzi A, Pacileo G, Limongelli G, Mingarelli R, Calabrò R, Marino B, Dallapiccola B. LEOPARD Sindrome: Clinical Diagnosis in the first Year of Life. American J Medical Genetics 140A; 740-746 (2006). PubMed.
9. Digilio MC, Sarkozy A, Pacileo G, Limongelli G, Marino B, Dallapiccola B. PTPN11 gene mutations: linking the Gln510Glu mutation to the "LEOPARD sindrome phenotype". Eur Pediatr (2006) 165; 803-805. PubMed
10. Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, van der Burgt I, Palleschi A, Petrucci TC, Sorcini M, Schoch C, Foà R, Emanuel PD, Gelb BD. Diversity and functional consequences of germline and somatic PTPN11 Mutations in human disease. The American J Human Genetics. Vol 78, Feb 2006, 279-290. PubMed
11. Kalidas K, Shaw AC, Crosby AH, Newbury-Ecob R, Greenhalgh L, Temple IK et all. Genetic heterogeneity in LEOPARD syndrome: two families with no mutations in PTPN11. J Hum Genet 2005, 50; 21-25. PubMed
12. Digilio MC, Sarkozy A, Pacileo G, Limongelli G, Conti E, Cerrato F, Marino B, Pizzuti A, Calabrò R, Dallapiccola B. Familiar Aggregation of genetically heterogeneous hypertrophic cardiomyopathy: a boy with LEOPARD sindrome due to PTPN11 mutation and his nonsyndromic father lacking PTPN11 mutations. Birth Defects Research (Part A) 70; 95-98 (2004). PubMed
13. Chong WS, Klanwarin W, Giam YC. Generalized Lentiginosis in Two Children Lacking Systemic Associations: Case Report and Review of the Literature. Pediatric Dermatology Vol. 21, N° 2, 139-145, 2004. PubMed
14. Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem 1997; 272: 32521-7. PubMed
15. Chang Y, Ceacareanu, Dixit M, Sreejayan N, Hassid A. Nitric oxide-induced motility in aortic smooth muscle cells; role of protein tyrosine phosphatase SHP-2 and GPT-binding protein Rho. Circ Res 2002; 91: 390-7. PubMed
16. Woywodt A, Welzel J, Haase H, Duerholz A, Wiegand U, Potratz J, Sheikhzadeh A. Cardiomyopathic Lentiginosis/LEOPARD Syndrome presenting as sudden cardiac arrest. Chest 1998, May 113(5). PubMed
© 2008 Dermatology Online Journal