Lamellar ichthyosis
Published Web Location
https://doi.org/10.5070/D34nt615vjMain Content
Lamellar ichthyosis
Frank Victor MD and Julie V Schaffer MD
Dermatology Online Journal 11 (4): 13
Department of Dermatology, New York University School of Medicine
Abstract
A 6-year-old African boy with a history of a collodion membrane presented with scale in a generalized distribution and flexural accentuation. Large, brown, polygonal scales were present on the forehead, lateral aspects of the face, and extremities. The nature of the scales and the lack of erythroderma in this patient are consistent with a mild form of lamellar ichthyosis (LI). LI and nonbullous congenital ichthyosiform erythroderma (NBCIE) represent phenotypes at the poles of the autosomal recessive ichthyosis spectrum. Mutations in genes encoding transglutaminase 1 (TGM1), the ABCA12 transporter (ABCA12), ichthyin, lipoxygenase 3 (ALOXE3), and 12(R)-lipoxygenase (ALOX12B) have been shown to underlie both NBCIE and LI.
A 6-year-old African boy presented to the Bellevue Hospital Center Pediatric Dermatology Clinic for evaluation of dry, scaly skin over his entire body. The patient was born at term encased in a collodion membrane. Over the first few weeks of life, the membrane was shed and replaced by scales in a generalized distribution. The patient spent the first 5 years of his life on the Ivory Coast and had occasionally experienced heat intolerance. He also had a history of painful fissures on the hands and feet. Petrolatum was applied on a daily basis, and emollient preparations containing urea and lactic acid had also been used.
An ophthalmologic examination and hearing evaluation disclosed no abnormalities. The patient had no other medical problems, and his growth and development have been normal. He was the first child of parents with no known consanguinity, and there was no family history of scaly skin.
Scale was present diffusely over the entire cutaneous surface with flexural accentuation. Larger, brown, polygonal scales were present on the forehead, lateral aspects of the face, and extremities, with prominent skin markings on the knees, ankles, and wrists. Finer scales were observed on the central face, neck, and trunk, with no apparent underlying erythema. The patient had hyperkeratotic, hyperlinear palms and soles. Facial skin was taut, but there was no ectropion, eclabium, or scarring alopecia. The nails were normal in appearance, and there was no hepatosplenomegaly.
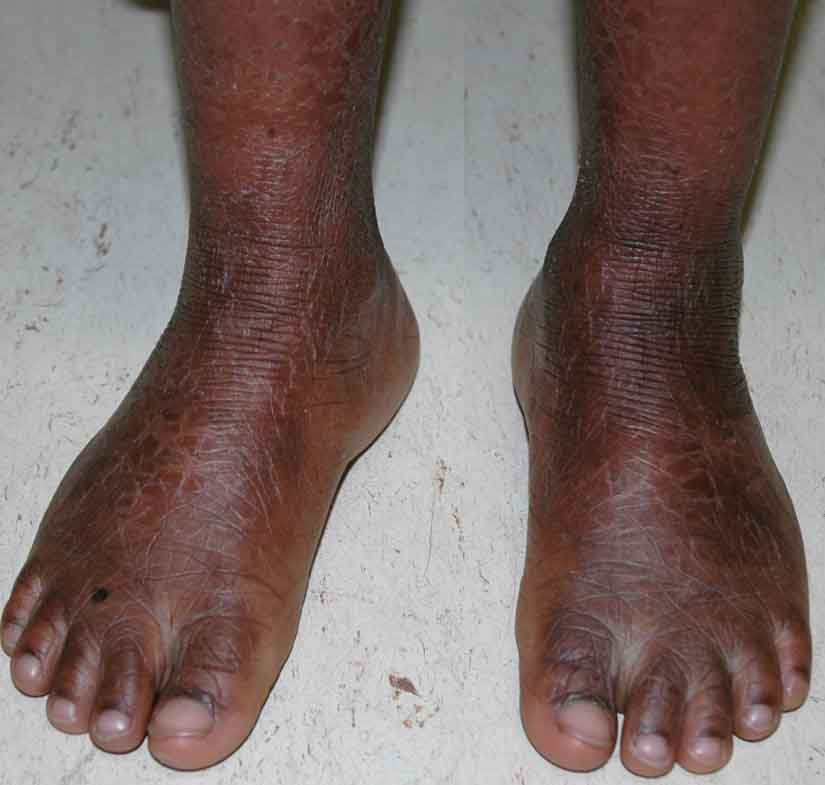
|

|
| Figure 1 | Figure 2 |
|---|
Examination of a peripheral blood smear showed no lipid vacuoles within granulocytes.
Comment
Lamellar ichthyosis (LI) is a genetically heterogeneous group of disorders of keratinization that are inherited in an autosomal recessive fashion. LI has an equal incidence in male and female individuals and is estimated to occur in approximately 1 in 300,000 live births. The large, dark, plate-like scales that characterize classic LI represent one end of the phenotypic spectrum of autosomal recessive congenital ichthyosis. The erythroderma and fine, white scales, which typify nonbullous congenital ichthyosiform erythroderma (NBCIE), lie at the opposite end of this spectrum, and various intermediate phenotypes have been observed [1]. Our patient's relatively mild form of LI, with smaller scales on the trunk than on the extremities and no erythrodermic component, could be considered as an intermediate phenotype that is closer to the LI than the NBCIE pole.
Mutations in the transglutaminase-1 (TGM1) gene on chromosome 14q11 account for approximately half the cases of LI and a minority of cases of NBCIE. The TGM1 mutations are heterogeneous (including point mutations, deletions, truncations, and splice-site mutations), and consistent genotype-phenotype correlations have not been observed [2, 3]. A lack of transglutaminase 1 activity impairs cross-linking of proteins and lipids in the cornified cell envelope of the upper epidermis and leads to defective cornification and desquamation [4].
In 2003 missense mutations in the ABCA12 gene on chromosome 2q35 were found to account for a LI phenotype in nine families from Northwest Africa [5]. As our patient originates from this geographical region, mutations in ABCA12 represent a potential explanation for his condition. Large deletions in ABCA12 were recently shown to cause harlequin ichthyosis (HI), a severe congenital skin disease that is usually lethal [6]. ABCA12 encodes a transmembrane transporter in the ATP-binding cassette family that is thought to play critical role in keratinocyte lipid trafficking and the formation of lamellar granules, which would explain the barrier defects observed in both LI and HI. Mutations in the genes encoding ichthyin, lipoxygenase 3 (ALOXE3), and 12(R)-lipoxygenase (ALOX12B) have also been shown to underlie both NBCIE and LI phenotypes.
Patients with LI are typically born encased in a translucent collodion membrane, which is replaced over the first month of life with generalized scale that is accentuated in flexural areas as well as on the forehead and lower extremities. Eclabium, ectropion, and scarring alopecia at the periphery of the scalp are often observed as a sequelae of excessively taut skin, and heat intolerance frequently occurs due to obstruction of the sweat ducts by plates of scale. Patients may also develop palmoplantar keratoderma, pseudoainhum, and nail dystrophy [1].
The use of emollients remains a cornerstone of treatment for LI. Although the use of keratolytics in children with LI may be limited due to skin irritation and the risk of systemic absorption, special formulations such as 5 percent lactic acid and 20 percent propylene glycol in a lipophilic base have been shown to be particularly effective in reducing scale. Topical retinoids and vitamin D3 derivatives may also be helpful, and topical application of the antioxidant N-acetylcysteine was reported to result in marked improvement in an adult woman with LI [7]. Oral retinoids are particularly effecting in reducing the amount of scale in LI patients, but this alteration may expose underlying erythroderma. Counseling patients with LI regarding overheating is important, and those with ectropion should receive ophthalmologic care [1]. Lastly, LI has been studied as a prototype for therapeutic cutaneous gene delivery. Restoration of tranglutaminase activity in keratinocytes from LI patients has been shown to normalize protein cross-linking and cornification in vitro and in a human skin/immunodeficient mouse xenograft model [8, 9].
References
1. Akiyama M, et al. The clinical spectrum of nonbullous congenital ichthyosiform erythroderma and lamellar ichthyosis. Clin Exp Dermatol 2003;28:2352. Candi E, et al. Transglutaminase 1 mutations in lamellar ichthyosis: loss of activity due to failure of activation by proteolytic processing. J Biol Chem 1998;273:13693
3. Hennies HC, et al. Genotype/phenotype correlation in autosomal recessive lamellar ichthyosis. Am J Hum Genet 1998;62:1052
4. Jeon S, et al. Inability of keratinocytes lacking their specific transglutaminase to form cross-linked envelopes: absence of envelopes as a simple diagnostic test for lamellar ichthyosis. Proc Natl Acad Sci 1998;95:687
5. Lefèvre C, et al. Mutations in the transporter ABCA12 are associatred with lamellar ichthyosis type 2. Hum Molec Genet 2003;12:2369
6. Kelsell DP, et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet 2005;76:794
7. Redondo P, et al. Topical N-acetylcysteine for lamellar ichthyosis. Lancet 1999; 354:1880
8. Choate KA, et al. Transglutaminase 1 delivery to lamellar ichthyosis keratinocytes. Hum Gene Ther 1996;7:2247
9. Choate KA, et al. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat Med 1996;2:1263
© 2005 Dermatology Online Journal