Primary, systemic amyloidosis and the dermatologist: Where classic skin lesions may provide the clue for early diagnosis
Published Web Location
https://doi.org/10.5070/D34kp7580wMain Content
Primary, systemic amyloidosis and the dermatologist: Where classic skin lesions may provide the clue for early diagnosis
Sophie R Silverstein MB BChir MA
Dermatology Online Journal 11 (1): 5
Whittington Hospital, London. sophiesilverstein@hotmail.com
Abstract
Primary amyloidosis may present with skin lesions as a primary or sole expression of underlying plasma-cell dyscrasia. Classic skin lesions of primary, systemic amyloidosis are listed, and features suggestive of the diagnosis are discussed. Where this condition is considered in the dermatologist's differential, the investigations described may lead to an early diagnosis. When therapy can be initiated before the onset of organ failure, survival may be prolonged.
Introduction
Systemic amyloidosis has well-recognized dermatological signs that may be the presenting features of the disease. Skin and soft-tissue lesions may, indeed, be the only manifestations of the disease prior to later-stage organ involvement, at which point treatment options are limited. Although the skin manifestations of systemic amyloid are common to many conditions, their presentation in certain clinical settings should help to indicate this disorder for inclusion in the differential diagnosis. With the appropriate investigations the dermatologist may thus have the opportunity to diagnose amyloidosis at an early stage.
Primary systemic amyloidosis (AL amyloidosis) is a plasma-cell dyscrasia of unknown cause. Immunoglobulin light chains, or fragments of light chains, produced by plasma-cell clones form extracellular amyloid fibrils. Amyloid deposition can occur in any organ in the body, causing features such as congestive cardiac failure, renal failure and hepatosplenomegaly, as well as skin lesions. Both sexes are affected by this disease, with onset most commonly from the sixth decade. Although recent advances in therapy are encouraging, the prognosis for primary amyloidosis remains poor.
Amyloidosis and skin lesions
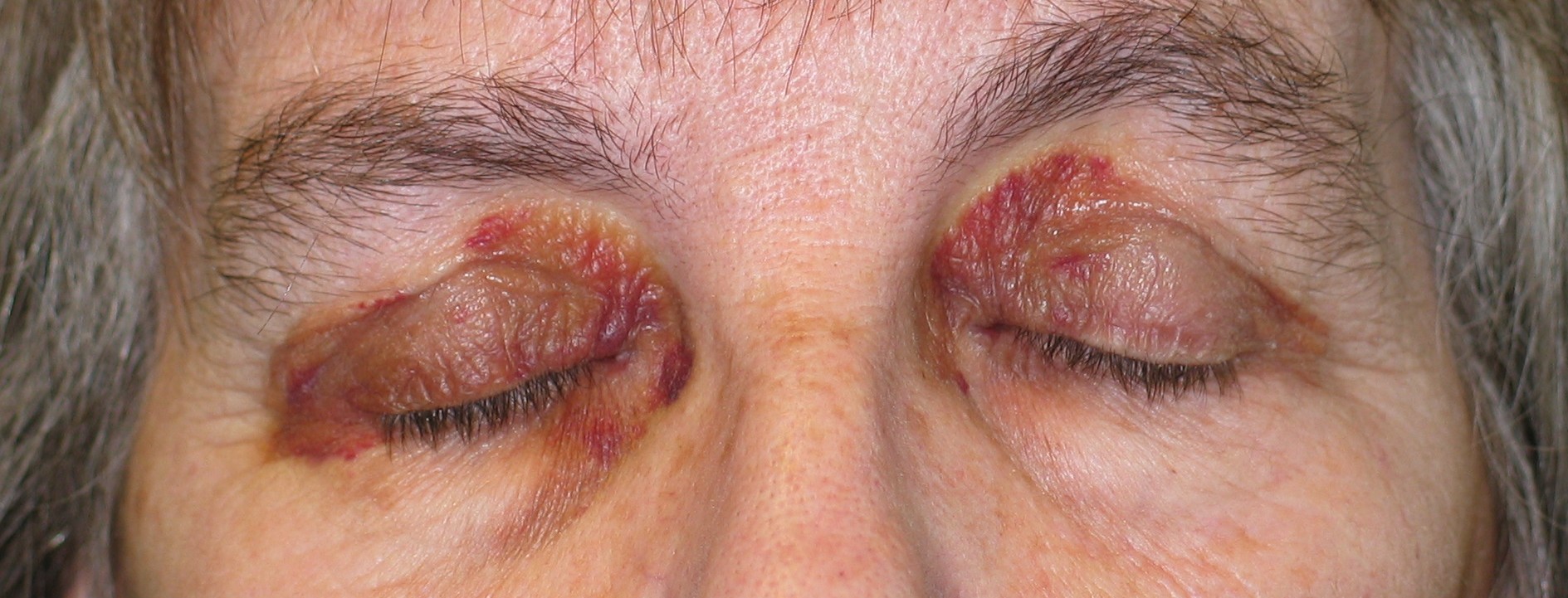
|
| Figure 1 |
|---|
| Periorbital purpura |
Amyloid deposition in the skin gives rise to particular dermatological lesions, which have been thoroughly described. Presentation to the dermatologist may be with any of the following features.
- Purpura, petechiae, and ecchymoses occur commonly in the skin and mucous membranes. They are due to intracutaneous hemorrhage, the result of amyloid infiltrating and weakening blood-vessel walls. These lesions may occur spontaneously and are particularly common in skin folds such as the eyelids, axillae, umbilicus, and anogenital area. Periorbital purpura may arise after coughing, sneezing, performing proctoscopy, or the valsalva maneuver [1] (Fig. 1).
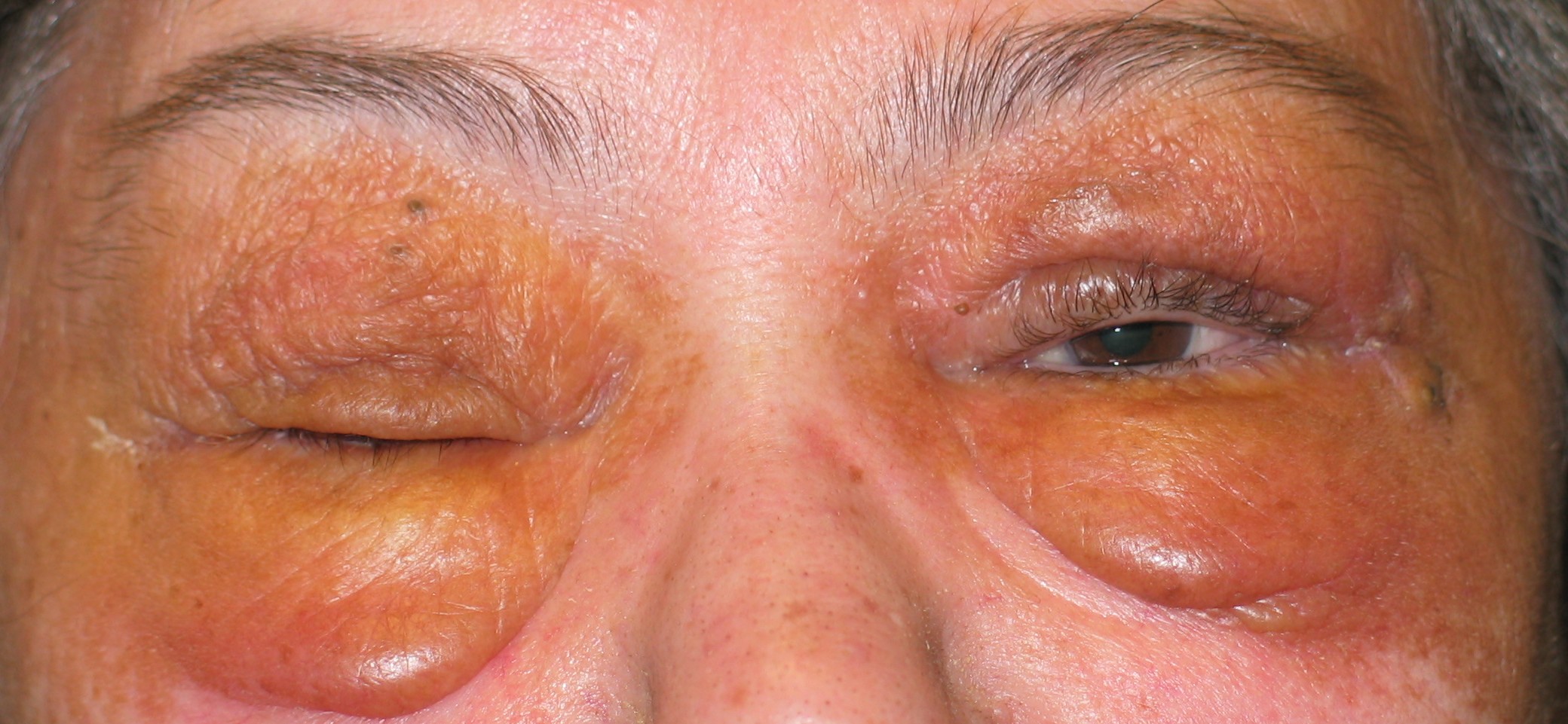
|
| Figure 2 |
|---|
| Waxy appearance of intradermal amyloid deposition around the eye. |
- Direct dermal infiltration can produce subcutaneous nodules and plaques. Classically described as smooth, waxy, yellowish lesions, these are uncommon and more often appear hemorrhagic. They are generally found on flexor surfaces, the face, and the buccal mucosa (Fig. 2). Cutaneous nodules can be demonstrated to follow the path of blood vessels within the skin [2]. Direct skin infiltration with amyloid can produce the appearance of scleroderma on the face, hands, and feet. Rarely, infiltrative nodular lesions may coalesce to form gross distortion and enlargement or tumefaction.
- Alopecia may develop, either in a patchy or in a uniform distribution.
- Nail dystrophy is not unusual in systemic amyloidosis. Features can include whitening of the nails, banding, brittleness, and onycholysis. There are reported instances of nail dystrophy being the only feature at presentation [3, 4].
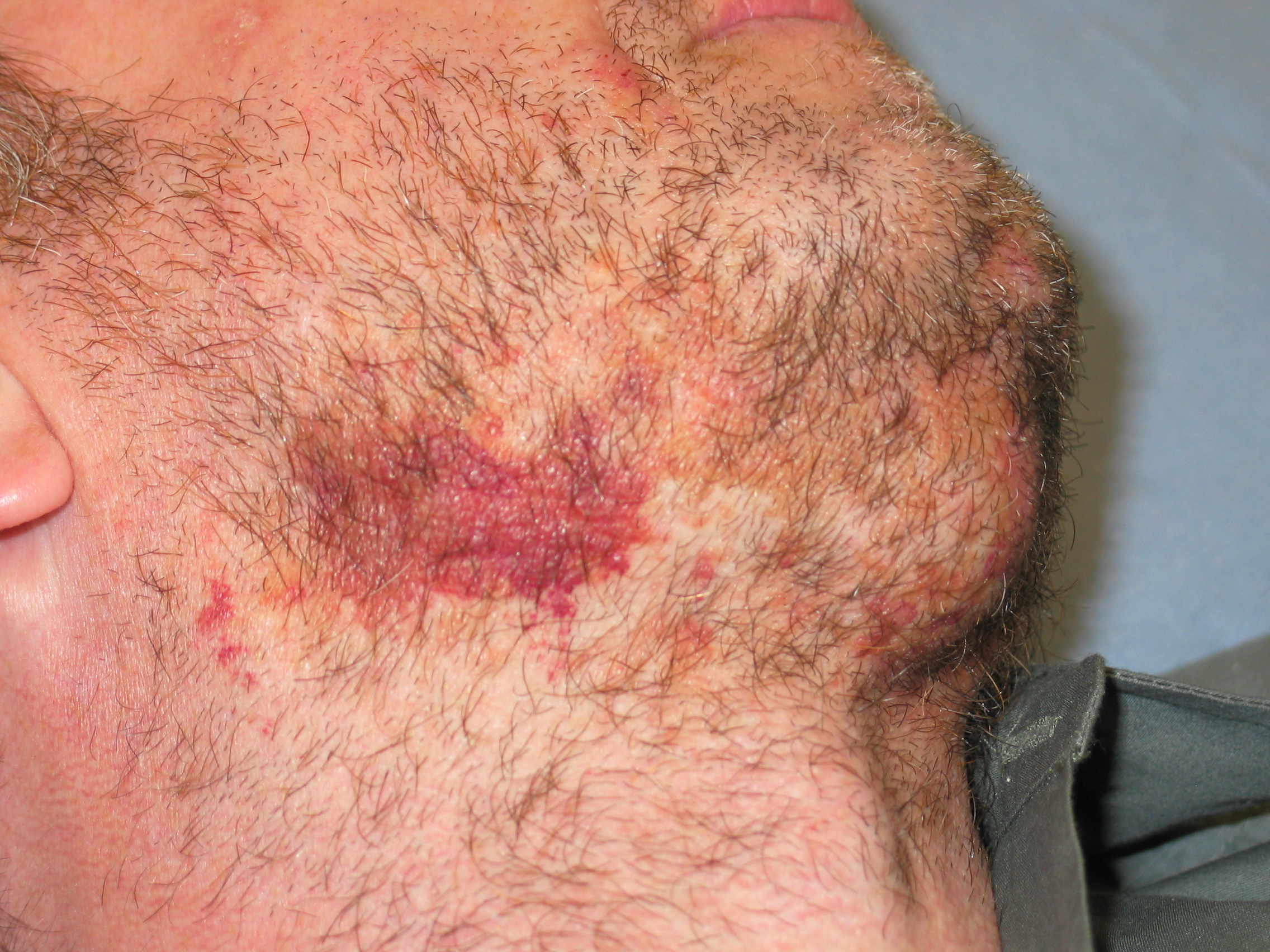
|
| Figure 3 |
|---|
| Hemorrhagic lesion over the mandible in a male patient, the result of shaving. There is also an enlarged submental lymph node. |
- Neuropathic skin changes may follow the infiltration of peripheral nerves with amyloid. Patients may present with traumatic lesions, ulcers, ecchymoses, and subcutaneous infection, secondary to peripheral neuropathy (Fig. 3).
- Skin discoloration can occur. Pallor, although usually associated with an underlying anemia, may occur in patients with a normal hemoglobin. Yellow discoloration of the skin may be attributed to underlying jaundice, or may be a feature of the waxy skin lesions described above. Jaundice and itching can be presenting features of amyloidosis and may follow hepatic amyloid deposition or else hemorrhage secondary to blood vessel infiltration.
- Rarely, bullae may form on the skin or mucous membranes. These may be confused with bullous pemphigoid, clinically and histologically [5].
- Target lesions have been described, arising around long-standing Campbell de Morgan spots.
When to consider amyloid
Type AL amyloidosis is rare, with an incidence of less than 1 per 100,000 in the United Kingdom, but mucocutaneous lesions may occur in up to 40 percent of these patients [6]. Among 223 new patients presenting to the Amyloid Treatment and Research Center in Boston during 1999, 28 percent had at least one of the aforementioned dermatological manifestations of AL amyloidosis. Crucially, skin lesions may be early or even the presenting feature of disease, and in some cases, soft tissue involvement may be the only manifestation of amyloidosis until later organ failure [7, 8]. Amyloidosis is a multisystem disease, and its presentation may mimic a wide range of conditions. Should the dermatologist be presented with early skin stigmata, there may be a valuable opportunity for early diagnosis should the disease be included appropriately in a working differential on initial consultation. Intervention may then be initiated before end-organ failure, allowing the possibility of a wider range of treatment options and greater opportunity to delay the development of disease. The greatest prognostic factor is the stage of disease at the time of initial treatment, demonstrating that early diagnosis is of paramount importance [9].
The consultation
Lesions such as purpura, alopecia, or ecchymoses may appear in a vast range of disorders seen by the dermatologist. Specific types of coincident disease may arouse suspicion of a rare cause of these common lesions. The presence of organ failure, particularly of renal or cardiac impairment, would be strong indication for further investigation for amyloid, although the patient may already be under investigation for such conditions. Careful history taking and examination may elicit early symptoms and signs of disease that would hint to the possibility of amyloidosis as a diagnosis, prompting key investigations for confirmation.
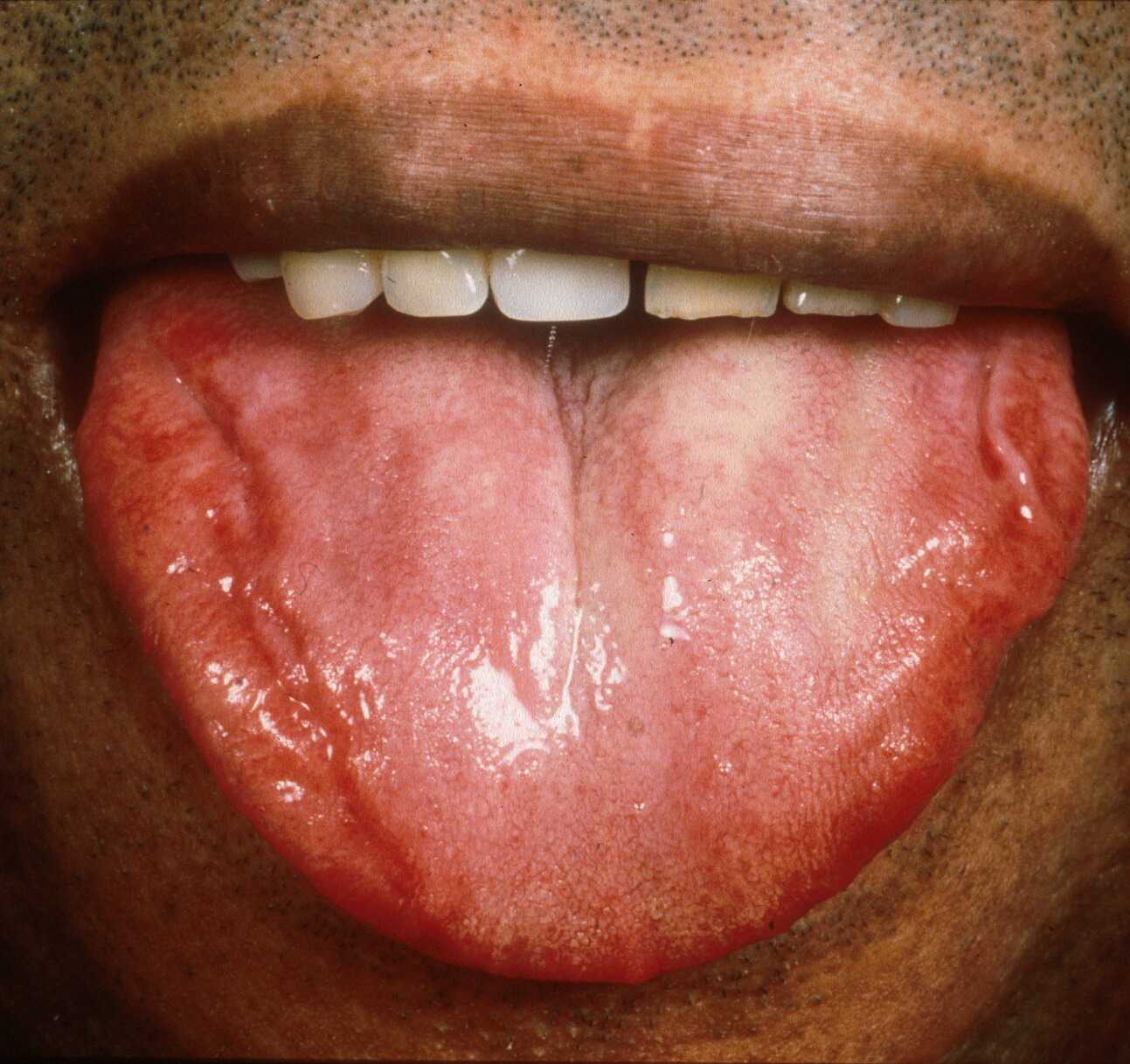
|
| Figure 4 |
|---|
| Macroglossia, which can be subtle |
Particularly suggestive would be a history of previous carpal tunnel syndrome or of symptoms of dysphagia, dysphonia, or macroglossia (Fig. 4). Any symptoms supporting cardiac or renal impairment would be relevant, including dyspnea, postural hypotension, and peripheral edema. Jaw and leg claudication can occur secondary to blood-vessel infiltration, which is also the mechanism responsible for bowel signs such as malabsorption and diarrhea. The patient may be short of breath, with altered performance status, or complain of fatigue, weight loss, anorexia, or lethargy.
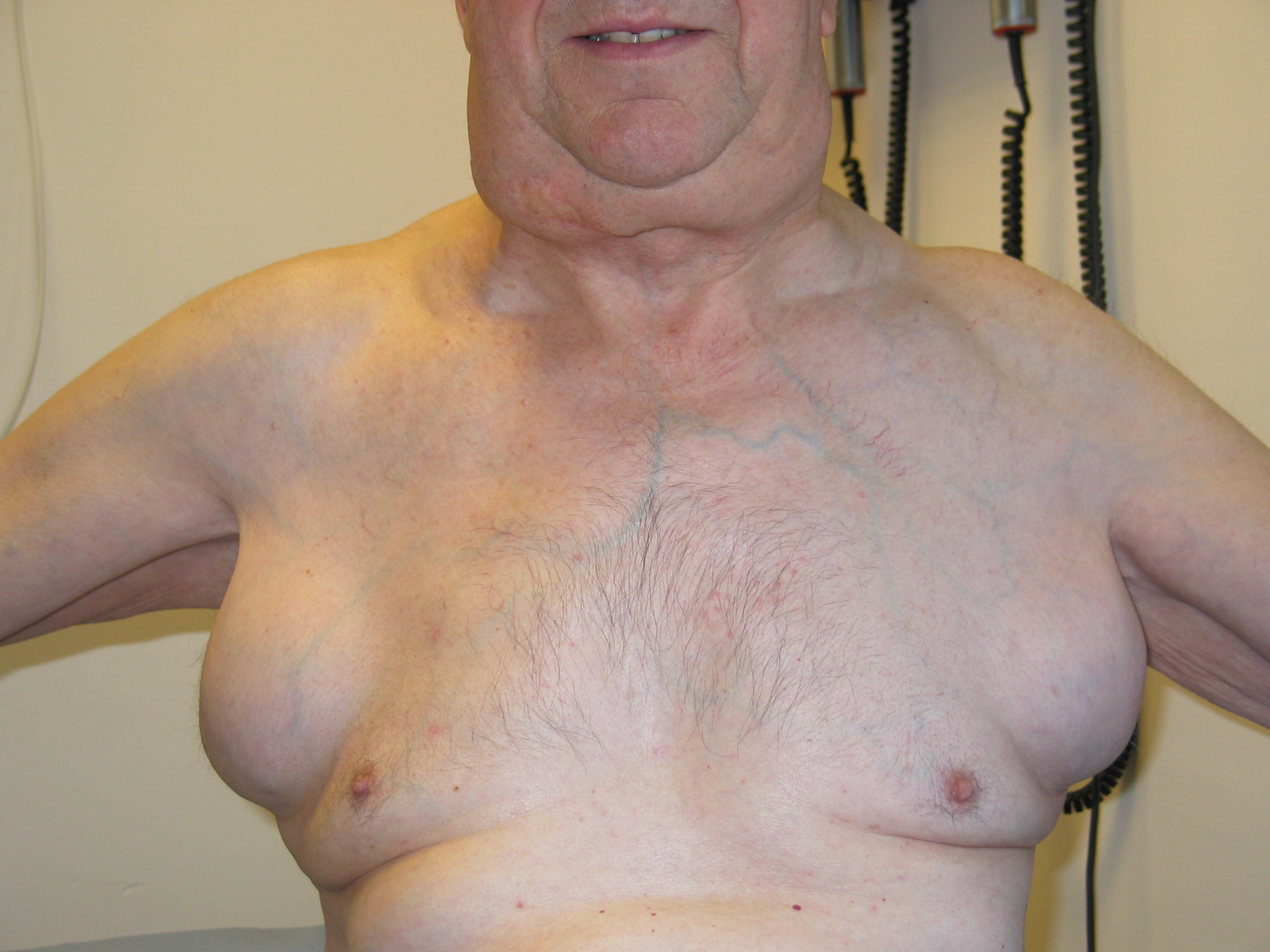
|
| Figure 5 |
|---|
| Gross lymphadenopathy |
On examination, the skin lesions listed above should be sought. There may be lymphadenopathy, which can be gross (Fig. 5). The most common organ affected by amyloid infiltration is the kidney; urinalysis may reveal proteinuria, a particularly useful side-room test because 80-90 percent of patients have proteinuria at some point. Blood pressure may reveal postural hypotension, commonly a feature of autonomic involvement. Hepatomegaly may be found in approximately 50 percent of patients; splenomegaly is less common. Occasionally amyloidosis may mimic features of rheumatoid arthritis, findings attributed to amyloid deposition in joints. Amyloidosis may also have features of Sjogren syndrome, attributed to direct dermal infiltration.
Investigations
Biopsy is the definitive investigation for amyloidosis. Fine-needle fat-pad biopsy and rectal biopsy have both been hailed as the investigation of choice, each giving positive results in 80-90 percent of cases [10, 11, 12]. Skin biopsy is also a useful investigation, with the possibility of providing definitive diagnosis. The biopsy may be of involved or uninvolved skin, as both provide high-yield results for diagnosis. Where other tissues are involved, they may also be used to stain for amyloid, including liver or spleen biopsy, carpal tunnel tissue, nail or scalp biopsy.
It is important to collect urine and serum samples for immunoelectrophoresis in order to demonstrate the presence of a circulating monoclonal protein. Bone-marrow biopsy is often performed at a later stage. Clotting studies should be performed, because coagulation abnormalities have been recognized in primary systemic amyloidosis. Routine liver-function tests, serum urea, electrolytes, a full blood count, and erythrocyte sedimentation rate should also be taken.
Results
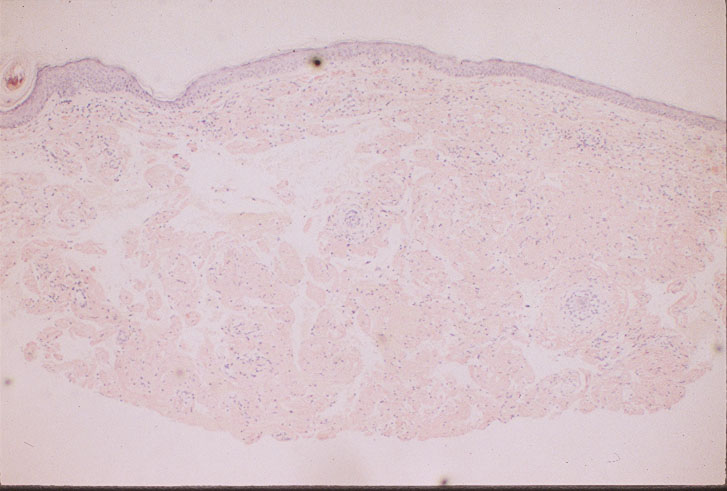
|
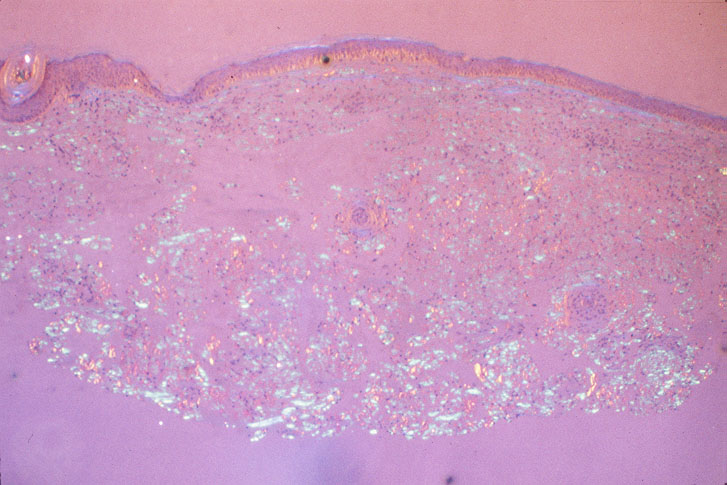
|
| Figure 6 | Figure 7 |
|---|---|
| Punch biopsy of skin stained with Congo red, demonstrating amyloid deposition (Fig. 6). | |
| View with polarizing light, accentuating amyloid deposits (Fig. 7). | |
On biopsy, amyloid can be seen as a homogenous, hyaline, fibrillary deposit. Congo-red staining is used to differentiate amyloid from other hyaline deposits (Fig. 6). On further treatment with potassium permanganate, AL amyloid retains its affinity for the red stain beneath a polarizing microscope, showing green birefringence, unlike the AA amyloid of systemic reactive amyloidosis (Fig. 7).
Skin biopsy characteristically shows diffuse amyloid deposition in the form of nodules and plaques. There may be amyloid infiltration of blood-vessel walls, around individual fat cells, and in pilosebaceous units. Amyloid deposits have also been identified in the nailfold and bed of dystrophic nails. These changes may also often be seen in biopsies of clinically normal skin. Distinction between systemic and localized forms of amyloid on skin biopsy does not usually present serious difficulty.
Blood tests may show a normal leukocyte and differential count. There will commonly be thrombocythemia, as well as clotting and liver-function test abnormalities. Approximately 50 percent of patients show an abnormal serum creatinine level, indicating a degree of renal insufficiency. Immunoelectrophoresis of both urine and plasma shows monoclonal protein in almost 90 percent patients, although this figure falls if only one of these specimens is examined [13]. Bone-marrow biopsy may show a modestly increased proportion of plasma cells or stain positively for amyloid. Staining of plasma cells for clonality may also be performed.
Treatment
The prognosis for AL amyloidosis is poor, with death usually resulting from cardiac or renal failure. Sudden death from cardiac arrhythmias also occurs. The mean survival from diagnosis is approximately 15 months [14]. Congestive cardiac failure is a particularly poor prognostic factor, and in many cases diagnosis at early stage may widen the therapeutic window [15]. Treatment is aimed at decreasing amyloid production and deposition and promoting lysis of deposits. Response to treatment may be complete, partial, or negative. Current treatment options include oral melphalan with or without prednisone, as well as stem-cell harvest and transfer, with intravenous melphalan or other chemotherapeutic agents. The use of dimethyl sulfoxide or colchicine has also been described, although with less success. Melphalan has been shown in trials to decrease new amyloid production and deposition, and to prolong survival [16]. Stem-cell harvest and transfer, after the use of either melphalan or other chemotherapeutic agents, has also been shown to improve survival [17]. Supportive measures include symptomatic treatment for organ impairment, such as diuretics for cardiac impairment, or hemodialysis in the case of renal failure. Cardiac, renal or liver transplantation may be carried out prior to stem cell transplantation where disease is isolated to a single organ [18, 19]. There are also case reports of organ transplantation used as rescue therapy or to improve organ function prior to stem-cell transplantation, although this remains controversial [20, 21].
Conclusion
Type AL amyloidosis is a rare condition with a poor prognosis. Presentation and even the sole manifestations of the disease may be skin lesions alone, so the dermatologist may have the opportunity to detect unrecognized cases of this disease. Where skin lesions follow classic patterns, specific elements of accompanying disease may give clues to amyloidosis as the potential etiology. With the appropriate tests, an early diagnosis of amyloidosis may be made and further organ involvement evaluated. As treatment outcome is dependent on stage at diagnosis, both treatment and survival are heavily influenced by the speed of recognition of the disease. Systemic amyloidosis has a wide array of pathological manifestations, which the dermatologist may have a vital role in recognizing.
With thanks to Dr R Gandour-Edwards of the University of California School of Medicine, for kindly allowing reproduction of slides. Also to Dr H Lachmann and the National Amyloidosis Centre, Royal Free and University College Medical School, for generously allowing use of their photographs.
References
1. Kyle RA, Bayrd ED: Amyloidosis: Review of 236 Cases. Medicine 1975 Jul; 54(4):271-99. PubMed2. Henry RB 3rd et al: Vascular amyloid in a patient with multiple myeloma. J Am Acad Dermatol 1986 Aug;15(2 Pt 2):379-82. PubMed
3. Fanti PA et al: Nail changes as the first sign of systemic amyloidosis. Dermatologia 1991;183(1):44-6. PubMed
4. Wheeler GE, Barrows GH: Alopecia Universalis. A manifestation of occult amyloidosis and multiple myeloma. Arch Dermatol 1981 Dec;117(12):815-6. PubMed
5. Johnson TM et al: Bullous amyloidosis. Cutis 1989 Apr;43(4):346-52. PubMed
6. Breathnach SM: Amyloid and amyloidosis. J Am Acad Dermatol 1988 Jan;18(1 Pt 1):1-16. PubMed
7. Breathnach SM et al: Systemic amyloidosis with an underlying lymphoproliferative disorder. Report of a case in which nail involvement was a presenting feature. Clin Exp Dermatol 1979 Dec;4(4):495-9. PubMed
8. Wheeler GE, Barrows GH: Alopecia Universalis. A manifestation of occult amyloidosis and multiple myeloma. Arch Dermatol 1981 Dec;117(12):815-6. PubMed
9. Moreau P et al: Prognostic factors for survival and response after high-dose therapy and autologous stem cell transplantation in systemic AL amyloidosis: a report on 21 patients: Br J Haematol 1998 Jun;101(4):766-9. PubMed
10. Duston MA et al: Diagnosis of amyloidosis by abdominal fat pad aspiration. Analysis of four years' experience. Am J Med 1987 Mar;82(3):412-4. PubMed
11. Libbey CA et al: Use of abdominal fat pad aspirate in the diagnosis of systemic amyloidosis. Arch Int Med 1983 Aug;143(8):1549-52. PubMed
12. Blum A, Sohar E: The diagnosis of amyloidosis. Ancillary procedures. Lancet 1962 Apr;1(7232):721-3. PubMed
13. Kyle RA, Bayrd ED: Amyloidosis: Review of 236 Cases. Medicine 1975 Jul; 54(4):271-99. PubMed
14. Kyle RA, Bayrd ED: Amyloidosis: Review of 236 Cases. Medicine 1975 Jul; 54(4):271-99. PubMed
15. Duston MA et al: Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum 1980 Jan;32(1):82-5. PubMed
16. Kyle RA et al: A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone and colchicine. N Engl J Med 1997 Apr;336(17):1202-7. PubMed
17. Comenzo RL et al: Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light chain) amyloidosis: survival and responses in 25 patients. Blood 1998 May;91(10):3662-70. PubMed
18. Comenzo RL: Primary Systemic Amyloidosis. Curr Treat Options Oncol. 2000 Apr:1(1):83-9. PubMed
19. Kumar KS, Lefkowitch J, Russo MW, Hesdorffer C, Kinkhabwala M, Kapur S, Emond JC, Brown RS Jr: Successful sequential liver and stem cell transplantation for hepatic failure due to primary AL amyloidosis. Gastroenterology. 2002 Jun;122(7):2026-31. PubMed
20. Mohty M, Albat B, Fegueux N, Rossi JF: Autologous peripheral blood stem cell transplantation following heart transplantation for primary systemic amyloidosis. Leuk Lymphoma. 2001 Mar;41(1-2):221-3. PubMed
21. Nowak G, Westermark P, Wernerson A, Herlenius G, Sletten K, Ericzon BG. Liver transplantation as rescue treatment in a patient with primary AL kappa amyloidosis. Transpl Int. 2000;13(2):92-7. PubMed
© 2005 Dermatology Online Journal