Oto-Rhino-Laryngology (ORL) tumor presentation in a case of systemic AA amyloidosis
Published Web Location
https://doi.org/10.5070/D348k7p142Main Content
Oto-Rhino-Laryngology (ORL) tumor presentation in a case of systemic AA amyloidosis
I Khoudri MD1, T Marcil MD1, I Elmeknassi MD1, M Kzadri PhD2, N Ismaili PhD2, Y Afifi PhD1, K Senouci PhD1, B Hassam PhD1
Dermatology Online Journal 15 (11): 6
1. Department of Dermatology, Ibn Sina University Hospital, Rabat, Morocco. ikhoudri@hotmail.com2. Department of ORL, Hôpital des spécialités, Rabat, Morocco
Abstract
A 64-year-old male with no underlying disease presented with the development of multiple skin nodules, loss of sensation in the extremities, hoarseness, macroglossia, and pain in the oral cavity. Direct laryngoscopy showed nodules involving the oral cavity, oropharynx, supraglottic region, and vocal cords. Biopsy from skin nodules showed amyloid deposits staining with Congo red. Immunohistochemical staining was used for AA protein and was positive. Biopsy from the oral floor was also positive for amyloid. Oto-Rhino-Laryngology (ORL) involvement has been reported in approximately 40 percent of AL amyloidosis patients, but does not appear to be frequent in AA amyloidosis. Cutaneous manifestations in AA amyloidosis are rare, although cases with lesions presenting as purpura are reported occasionally; we are not aware of other cases of ORL nodular involvement in systemic AA.
Introduction
Amyloidosis is a heterogeneous group of disorders characterized by extracellular deposition of insoluble protein fibrils in various tissues and organs [1]. Deposition of amyloid can be localized in a single organ, or systemic in various organs and tissues throughout the body. The various clinical pictures of systemic amyloidosis are related to the types of the precursor protein involved. In the recent classification of amyloid precursor protein, up to 24 different proteins have been recognized to date to be amyloidogenic in humans [2]. Systemic amyloidosis is an uncommon disease that can involve many organ systems. There are two major forms of acquired systemic amyloidosis: AL amyloidosis, in which the fibrils are derived from immunoglobulin light chains, and AA amyloidosis, in which they originate from serum amyloid protein A [3].
Oto-Rhino-Laryngology (ORL) involvement is identical in all forms of amyloidosis, but has been reported in approximately 40 percent of AL amyloidosis patients and does not appear to be frequent in AA amyloidosis [4]. Cutaneous manifestations in AA amyloidosis are rare, although cases with purpura are reported occasionally [5, 6]. To our knowledge, a diffuse nodular skin picture in systemic AA amyloidosis has never been reported.
Case report
 |  |
| Figure 1 | Figure 2 |
|---|---|
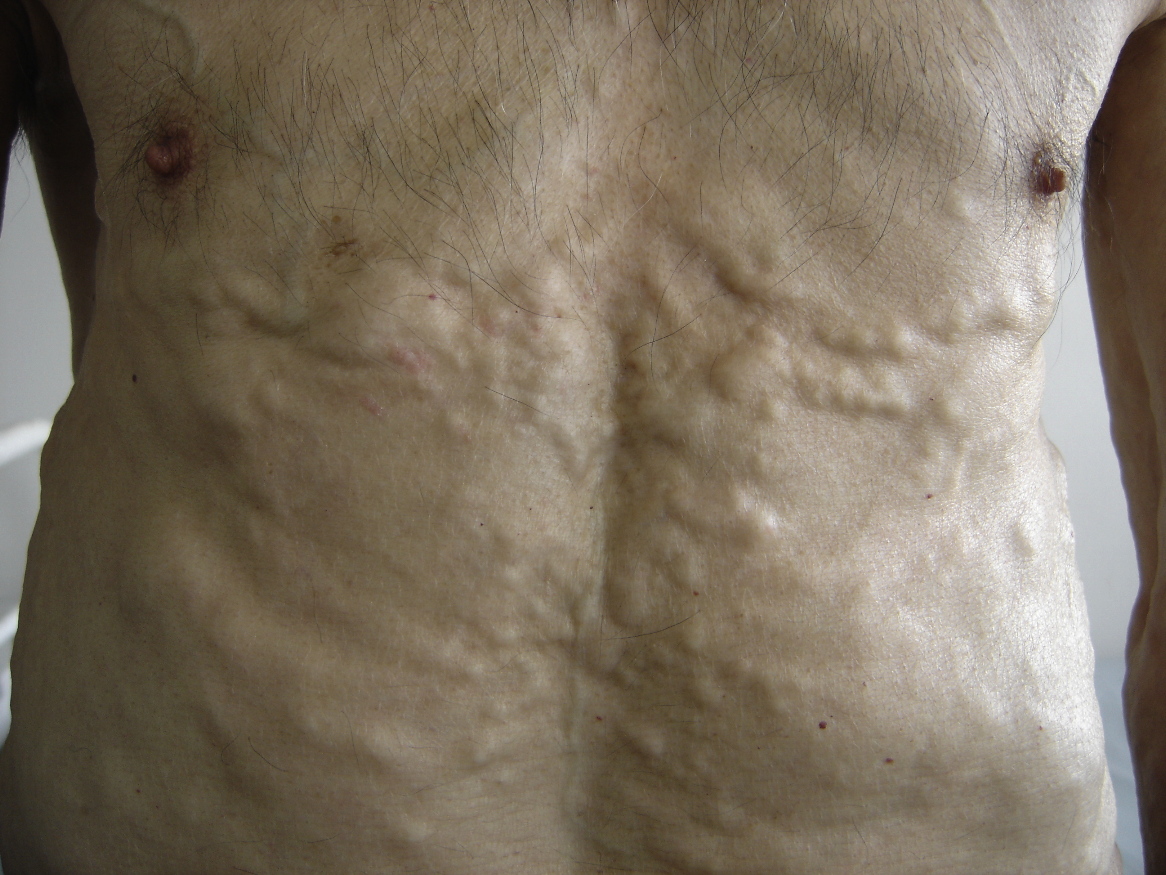
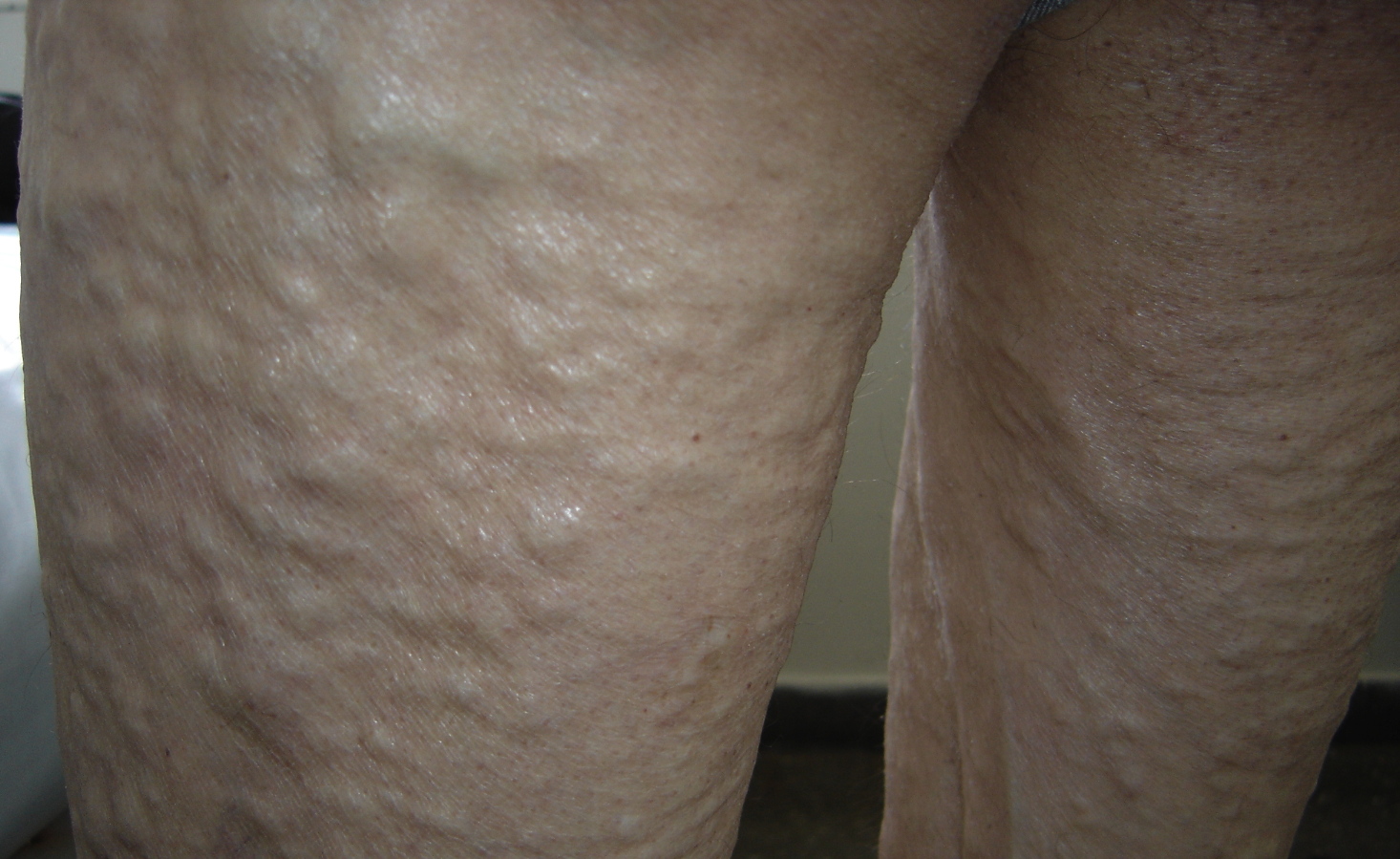
| Figure 1. Multiple skin-colored nodules of the trunk Figure 2. Multiple skin-colored nodules of the legs | |
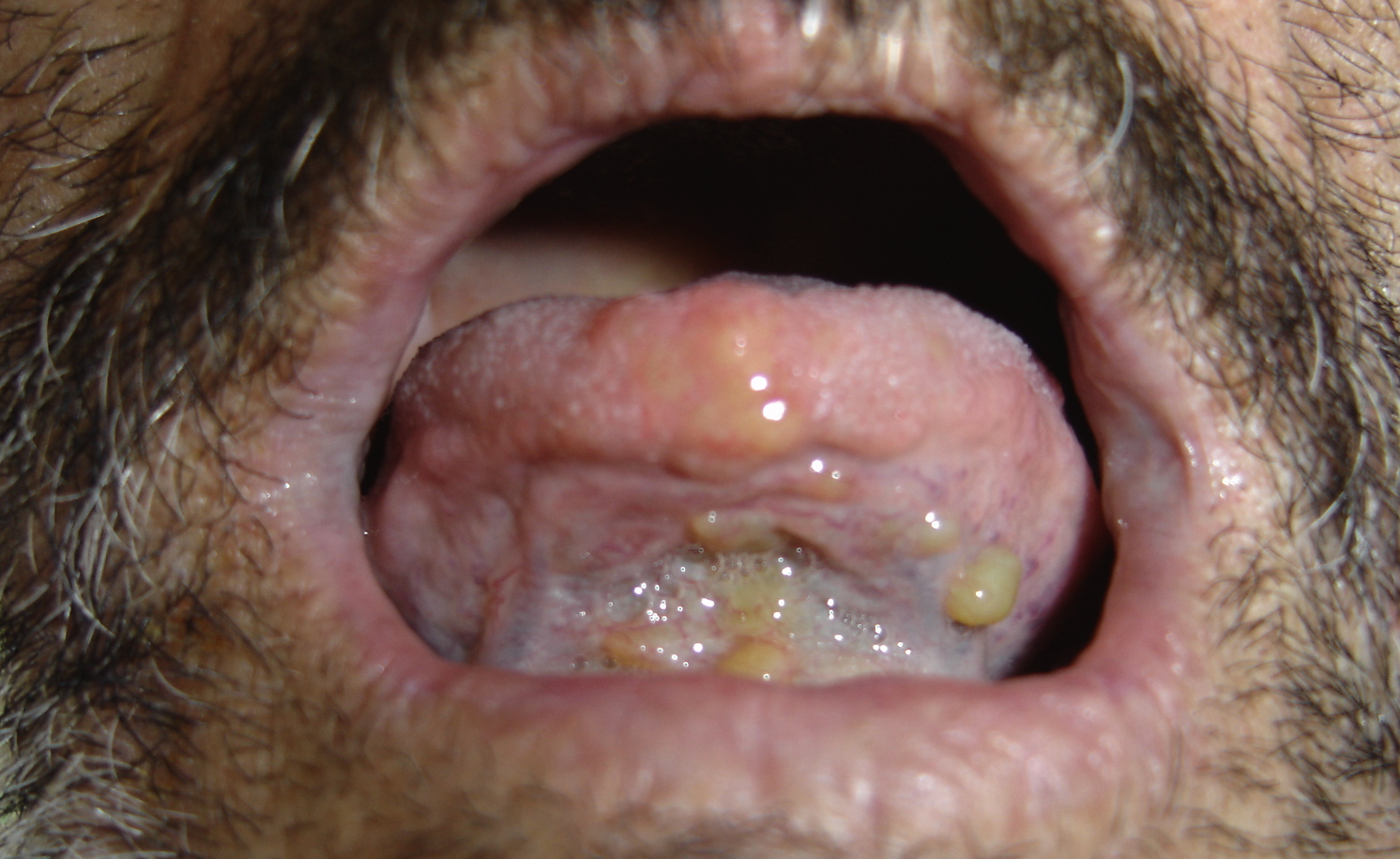
The patient was a 64-year-old male with no family history of underlying chronic inflammatory or infectious conditions such as rheumatoid arthritis. He presented a four-year history of multiple spontaneously developing skin nodules, loss of sensation and muscle strength of extremities, weight loss, weakness, and incapability of motion. Moreover, the patient was complaining of hoarseness, pain in the oral cavity, and difficulty of chewing and swallowing solid foods. Clinically, multiple firm and skin-colored nodules (Figs. 1 & 2) were found overlying the trunk, limbs, face, and neck. Oto-Rhino-Laryngology examination revealed macroglossia, a few yellow papules (Fig. 3), a tumorous appearance of the oral floor, and significant hypopharyngeal edema upon indirect laryngoscopy. Palpation showed no lymph node enlargement. Systemic examination revealed hypoesthesia of extremities, and abolished deep tendon reflex. Blood counts, serum biochemistry, and biological inflammatory markers were normal. Bacilloscopy was negative.
 |  |
| Figure 3 | Figure 4 |
|---|---|
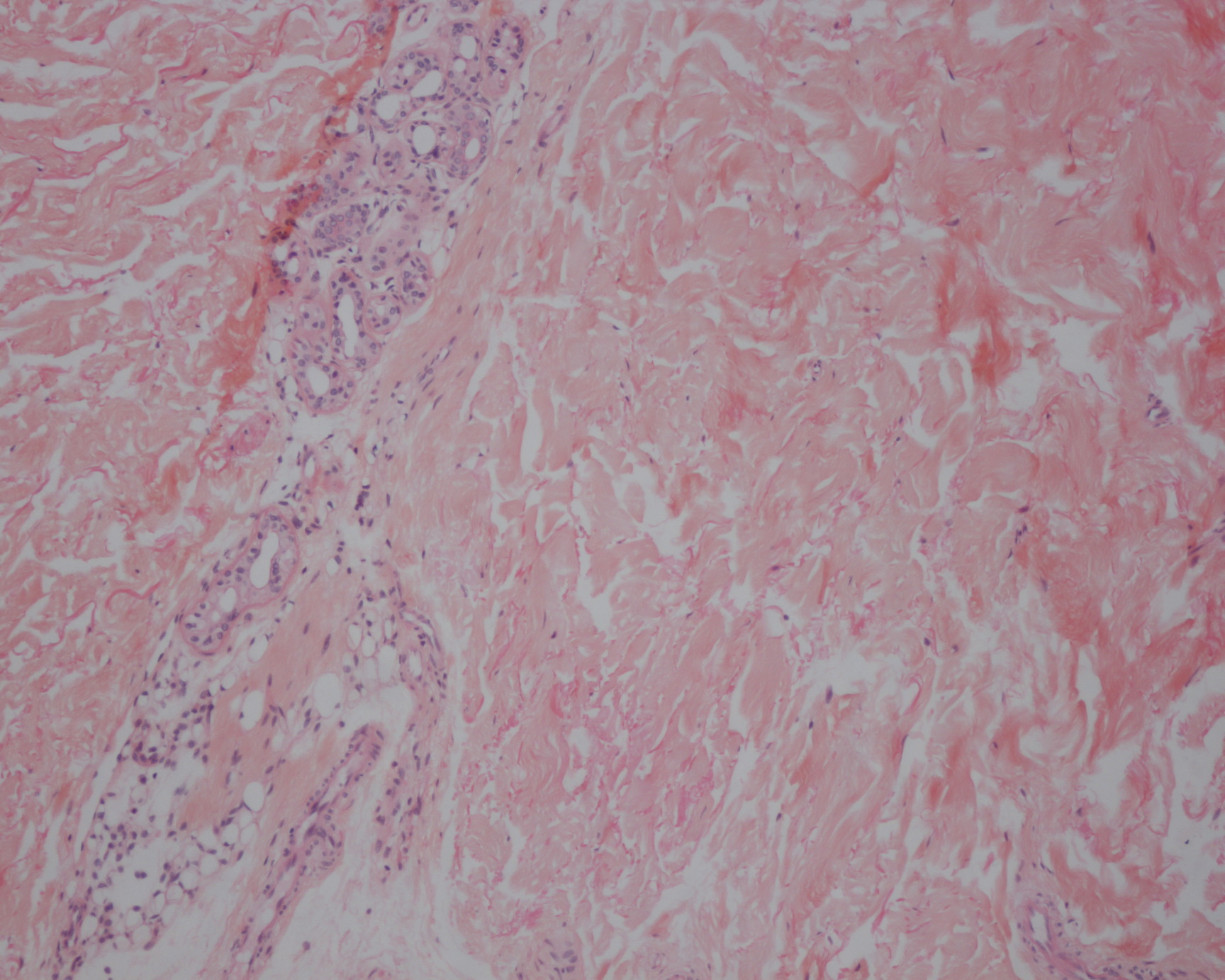
| Figure 3. Papular yellow deposits of the oral floor Figure 4. Microscopic examination with Congo red staining of a skin biopsy showing homogeneously red deposits of amyloid | |
Biopsy from skin nodules showed normal epidermis and eosinophilic nodules in the deep reticular dermis (Fig. 4). Staining with Congo red showed amyloid deposits with the characteristic apple-green birefringence in polarized light microscope. Loss of affinity for Congo red after pretreatment with potassium permanganate was noted. To check for systemic involvement, a second biopsy from the oral floor was also positive for amyloid. Cervical scan revealed edematous infiltration of the hypopharynx. Direct laryngoscopy showed amyloid deposits involving the oral cavity, oropharynx, supraglottic region, and vocal cords. Urine sediment and proteinuria were negative. Electrocardiography, echocardiography, and thoraco-abdominal scan were normal. To assess peripheral neuropathy, electromyography was not conclusive because of the infiltrated skin lesions. Serum and urine immunoelectrophoresis was negative, and bone marrow aspirate was normal, ruling out plasma cell dyscrasia.
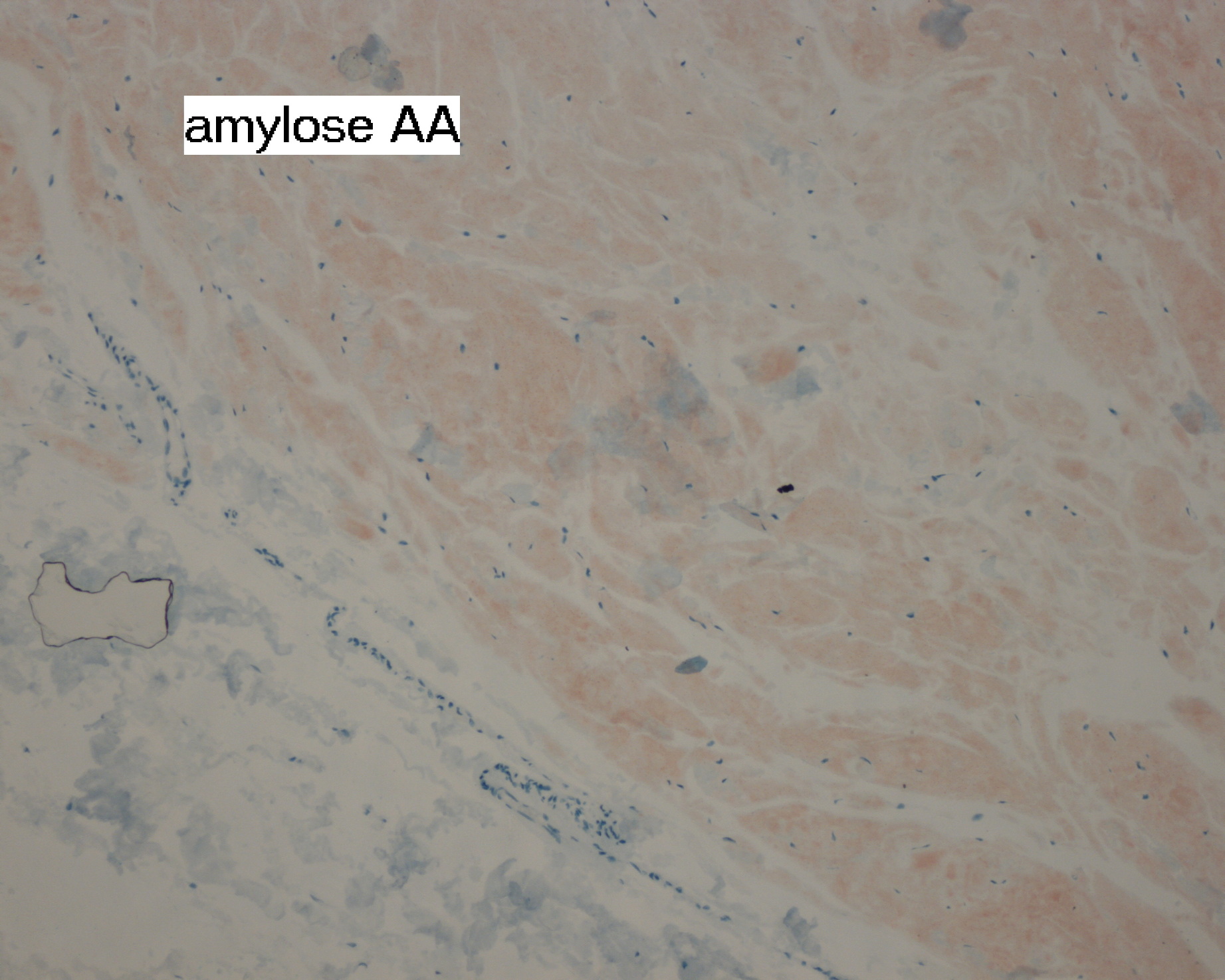 |
| Figure 5 |
|---|
| Figure 5. Immunihistochemistry of a skin biopsy using monoclonal antibodies against SAA (Reu.86.2). Amyloid is brown. |
Because no clinical signs or symptoms were evident to type the systemic amyloid, immunohistochemical staining of skin biopsy was used for AA protein and was positive showing brown deposits of amyloid in the immunoperoxydase stain with monoclonal antibodies against SAA (Reu.86.2) (Fig. 5). Amyloid A protein quantification was not performed. The patient was treated with colchicine, and no ORL intervention was required because of the lack of airway obstruction signs. The patient was dead three months later of unknown causes.
Discussion
Amyloidosis is an acquired or inherited disorder of protein folding [7]. AA amyloidosis is a less common form of systemic amyloidosis in comparison to AL amyloidosis, in which the incidence is estimated to be 0.8 per 100,000 person year [8]. AA amyloid, derived from the acute phase reactant serum amyloid A protein, has a lifetime incidence of 1-5 percent among patients with chronic inflammatory or infectious disorders [7]. It is then a rare complication of persistent inflammatory disease. Our patient developed a systemic AA amyloidosis without any known history of underlying inflammatory condition. This finding is not surprising; Ding-Dar et al. [3] reported a group of 20 cases of systemic amyloidosis that included three patients diagnosed with AA amyloidosis of which two patients were without known chronic inflammatory or infectious diseases.
The amyloid deposits were typed AA in our patient for two reasons. First, because the loss of affinity for Congo red after pretreatment with potassium permanganate is characteristic of AA amyloidosis [3]. Second, immunohistochemistry staining with antibodies to AA protein is a more reliable method to distinguish AA from AL amyoidosis [3]. In the skin biopsy of our patient, amyloid was brown in the immunoperoxydase stain with monoclonal antibodies against SAA (Reu.86.2). Although amyloid A protein quantification could not be used in our case, immunohistochemistry of a biopsy is sufficient in AA amyloidosis when sensitive and specific antibodies are used such as Reu.86.2 and Reu.86.5 [9, 10]. Specific antibodies for AL amyloidosis were not used in our case, because this method using polyclonal antibodies is less reliable than in AA amyloidosis [11]. This is caused by heterogeneity of amyloid deposits, lower sensitivity and specificity of antibodies, and non-specific adherence of immunoglobulins to amyloid deposits [11]. AL amyloidosis can be demonstrated by the presence of lambda or kappa light chains in serum or urine (by immunofixation electrophoresis), by the presence in the bone marrow of a relative excess of cells producing one of the light chains, and by the lack of reactivity with anti-AA antibodies [9]. No sign of monoclonal cell dyscrasia was noted in serum, urine, and bone marrow of our patient.
In general, AA amyloid deposits first occur in the spleen and liver and may long remain asymptomatic; the kidney is most often affected causing proteinuria, nephrotic syndrome and progressive development of renal insufficiency. Other clinical consequences like peripheral neuropathy, goiter, and cardiac and gastrointestinal manifestations remain rare (10-20%) [7, 8]. The clinical presentation of our patient seems curious because systemic amyloidosis was microscopically confirmed in two sites (skin and ORL) and he probably had amyloid-related peripheral neuropathy, but no symptomatic renal involvement. Moreover, cutaneous manifestations in AA amyoidosis are exceptional; purpura has been occasionally reported [5, 6]. The skin is frequently affected in localized amyloidosis [11]. Thus, our patient with multiple cutaneous and mucosal nodules skin lesions revealing a systemic AA amyloidosis is highly unusual. Histopathologically, Li [12] proposed that if deeper amyloid deposits are found in a biopsy of cutaneous amyloidosis, the possibility of systemic amyloidosis should be considered and further investigations should be performed. These investigations were done in our patient.
"Senile systemic amyloidosis" is a hereditary amyloidosis characterized by precursor protein transthyretin (TTR) mutation; it is seen in elderly patients and can lead to congestive heart failure. Sporadic cases with peripheral neuropathy and presence of a TTR mutation have been described [13]. Our patient had an older age with an unusual clinical presentation and no known underlying disease. Although genetic testing was not used in our case, immunohistochemistry is sufficient to confirm the AA amyloidosis type and eliminate the "senile systemic amyloidosis" hypothesis because of the high sensitivity and specificity of monoclonal antibodies [11]. Otherwise, the chance association of a monoclonal gammapathy in an older patient with systemic amyloidosis must be kept in mind. In our case, no evidence of monoclonal gammapathy was found.
Median survival of systemic AA amyloidosis after diagnosis is 4-10 years [8]. Death occurrs by destroying the normal structure and function of organs. Treatment is aimed at decreasing SAA protein by using cytostatic drugs, anti-TNF drugs, and a promising new drug Fibrillex® [11]. Colchicine has a central place in the treatment of familial Mediterranean fever, but was the only possible therapeutic modality for our patient taking into account the socio-economic conditions.
Amyloidosis in the ORL field is relatively rare. The larynx is the most common site of involvement and occurs largely in AL amyloidosis [14, 15]. Those patients usually present with long-standing hoarseness or dyspnea. Oto-Rhino-Laryngology amyloidosis is usually localized and the possibility of systemic disease is rare [4]. Bartels et al. [16] reported 188 patients with amyloidosis and only six of these had localized laryngeal AL amyloidosis; the sixth patient turned out to have systemic AL amyloidosis 8 years later. Oto-Rhino-Laryngology lesions may then represent the initial manifestation of AL amyloidosis or the first sign of monoclonal gammapathy. Otherwise, ORL tumorous appearance in systemic AA amyloidosis is exceptional; macroglossia and oral nodular deposits are even more rare in AA amyloidosis [4]. Buccal mucosa and salivary glands may also be affected. Treatment of ORL amyloid deposits is a complex problem because the papules and nodules frequently recur and repeated excisions are required [4]. The first line of treatment should include carbon dioxide laser microsurgery or cold endoscopic excision. Surgery remains the treatment of choice, but voice and airway preservation should be the aim in all patients [14, 15, 16].
In conclusion, this case report of systemic AA amyloidosis is peculiar by the lack of chronic inflammatory disease or multiple nodular skin lesions. In addition, our patient had an unusual tumor ORL presentation. This case highlights the heterogeneity of amyloidosis in clinical manifestations and in the pattern of amyloid-associated organ toxicity.
References
1. Husby G. Nomenclature and classification of amyloid and amyloidosis. J Intern Med 1992; 232: 511-2 [PubMed]2. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349: 583-96 [PubMed]
3. Lee DD, Huang CY, Wong CK. Dermatopathologic Findings in 20 Cases of Systemic Amyloidosis. Am J Dermatopathol 1998; 20: 438-42 [PubMed]
4. Koloktronis A, Chatzigiannis I, Paloukidou N. Oral involvement in a case of AA amyloidosis. Oral Dis 2003; 9: 269-72 [PubMed]
5. Libbey CA, Skinner M, Cohen AS. Use of abdominal fat tissue aspirate in the diagnosis of systemic amyloidosis. Arch Int Med 1993; 143: 1549-52 [PubMed]
6. Rubinow A, Cohen AS. Skin involvement in generalized amyloidosis. A study of clinically involved and uninvolved skin in 50 patients with primary and secondary amyloidosis. Ann Intern Med 1978; 88: 781-5 [PubMed]
7. Hirschfield GM. Amyloidosis: a clinico-pathophysiological synopsis. Semin Cell Dev Bio. 2004; 15: 39-44 [PubMed]
8. Obici L, Perfetti V, Palladini G, Moratti R, Merlini G. Clinical aspects of systemic amyloid diseases. Biochim Biophys Acta 2005; 1753: 11-22 [PubMed]
9. Hazenberg BP, Bijzet J, Limburg PC, Skinner M, Hawkins PN, Butrimiene I, et al. Diagnostic performance of amyloid A protein quantification in fat tissue of patients with clinical AA amyloidosis. Amyloid 2007; 14: 133-40 [PubMed]
10. Hazenberg BP, Limburg PC, Bijzet J, Rijswijk MH. A quantitative method for detecting deposits of amyloid A protein in aspirated fat tissue of patients with arthritis. Ann Rheum Dis 1999; 58:96-102 [PubMed ]
11. Hazenberg BP, van Gameren II, Bijzet J, Jager PL, van Rijswijk MH. Diagnostic and therapeutic approach of systemic amyloidosis. Neth J Med 2004; 62: 121-8 [PubMed]
12. Li WM. Histopathology of primary cutaneous amyloidosis and systemic amyloidosis. Clin Dermatol 1990; 8: 30-5 [PubMed]
13. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346: 1786-91 [PubMed]
14. Vázquez de la Iglesia F, Sánchez Ferrándis N, Rey Martínez J, Ruba San Miguel D, Rama López J, Fernández González S. Amyloidosis in the ORL field. Acta Otorrinolaringol Esp 2006; 57: 145-8 [PubMed]
15. Siddachari RC, Chaukar DA, Pramesh CS, Naresh KN, de Souza CE, Dcruz AK. Laryngeal amyloidosis. J Otolaryngol 2005; 34: 60-3 [PubMed]
16. Bartels H, Dikkers FG, van der Wal JE, Lokhorst HM, Hazenberg BP. Laryngeal amyloidosis: localized versus systemic disease and update on diagnosis and therapy. Ann Otol Rhinol Laryngol 2004; 113: 741-8. [PubMed]
© 2009 Dermatology Online Journal