Schwannoma of the eyelid: report of two cases
Published Web Location
https://doi.org/10.5070/D33s09x4knMain Content
Schwannoma of the eyelid: report of two cases
Elena López-Tizón MD1, Enrique Mencía-Gutiérrez MD1, Esperanza Gutiérrez-Díaz MD1, José R Ricoy MD PhD2Affiliations;Departments of Ophthalmology1 and Pathology2, 12 de Octubre Hospital, Complutense University, Madrid - Spain
Dermatology Online Journal 13 (2): 12
Abstract
Schwannomas are rare benign neurogenic tumors that show differentiation of Schwann cells that form the neural sheath. Only five reports of eyelid schwannomas in adults have been found in the English literature. We report the unusual cases of two females, aged 41 and 70 years, who developed eyelid schwannomas. Neither tumor was diagnosed clinically; both were erroneously considered as epidermal inclusion cysts. The masses were surgically removed by excisional biopsy. The histopathological examination showed encapsulated tumors composed of interlacing bundles of spindle cells with slightly wavy nuclei. Immunocytochemistry for S-100 protein was strongly positive. The diagnosis of schwannoma was made. After 12 and 2 years of follow-up, no recurrences have been observed. This entity should be included in the differential diagnosis of eyelid tumors.
Schwannomas (neurilemmomas) are benign tumors of neuroectodermic origin derived from the cells of Schwann that form the neural sheath. These tumors preferentially involve spinal nerve roots, and the sympathetic, cervical, and vagus nerves. They may be associated with neurofibromatosis, but solitary schwannoma at any site is not usually associated with this entity. Schwannomas have been reported in relation to the orbit, and, infrequently, to the uveal tract and conjunctiva. On the eyelid they are extremely uncommon; only five cases in adults [1, 2, 3, 4, 5] and one in a child [6] have been described.
Clinical synopsis
CASE 1
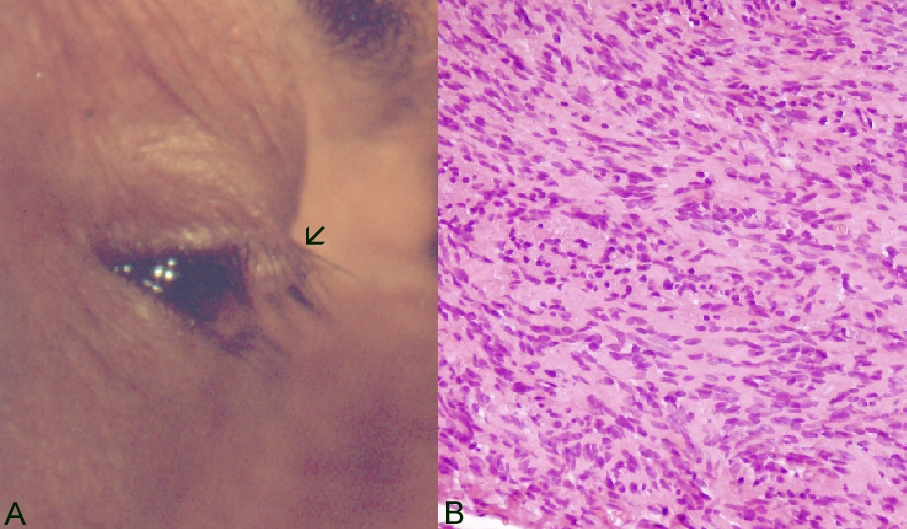
A 41-year-old female presented with a slowly enlarging, firm, 0.4 cm nodule located on the right upper eyelid margin (Fig. 1A). She had had the mass for 1 year and had no history of neurofibromatosis or any other nodules. The lesion was initially thought to be an inclusion cyst and was surgically treated by means of a pentagonal full-thickness excision. Macroscopically, the specimen was an apparently encapsulated smooth nodule.
Histologically the tumor was formed by fusiform cells arranged in intertwined bundles. The nuclei were fusiform and tended to form palisades (Fig. 1B). There was no mitotic activity. Immunochemistry for S-100 protein was strongly positive. These findings established the diagnosis of schwannoma. The patient recovered uneventfully and, after 12 years of follow-up, no evidence exists of local recurrence.
CASE 2
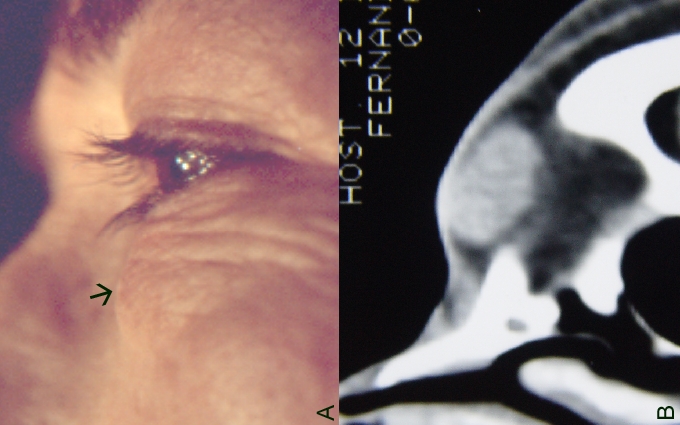
A 70-year-old female presented with a firm-nodular lesion, 1.7 cm in diameter, located on the middle of the left lower eyelid (Fig. 2A). The mass had a cystic appearance and cartilaginous consistence; it assumed a subcutaneous position and was mobile under the skin without attachment to deeper tissues. Computed tomography scan documented a nodular oval, well-circumscribed, solid-cystic lesion (Fig. 2B). Physical examination was unremarkable and no signs of neurofibromatosis were present. The lesion was initially thought to be an inclusion cyst and it was surgically excised by means of an anterior approach through the skin. Gross histopathological examination showed an encapsulated mass.
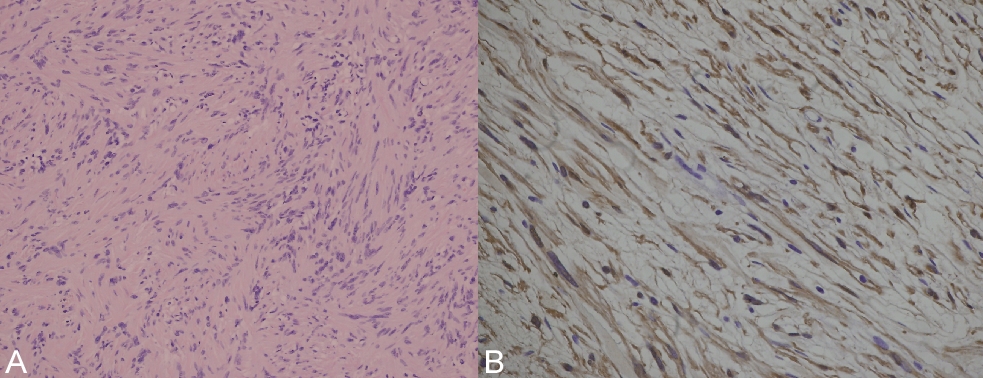
Microscopically, a lesion composed of fusiform cells arranged in intertwined bundles was revealed. The nuclei were oval and formed palisades. There was no mitotic activity (Fig. 3A). By immunocytohistochemistry techniques, the cells showed a strong positive reaction for S-100 protein (Fig. 3B). A diagnosis of schwannoma was made. After 2 years of follow-up, no evidence exists of local recurrence.
 |  |
| Figure 1 | Figure 2 |
|---|---|
| Figure 1. a) Case 1. Clinical appearance of a circumscribed schwannoma located on the edge in the middle part of the right upper eyelid in a 41-year-old woman (arrow). b) Gross histological features were fusiform cells with elongated nuclei (hematoxylin-eosin, original magnification x40). | |
| Figure 2. a) Case 2. Clinical appearance of schwannoma in the medial aspect of the left lower eyelid in a 70-year-old woman. Note the mass as subcutaneous (arrow). b) CT scan showed a well-circumscribed, solid-cystic lesion of 1.7 cm in diameter. | |
 |
| Figure 3 |
|---|
| Figure 3. a) Case 2. General aspect of lesion, constituted by fusiform cells with regular nuclei and scattered palisading (hematoxylin-eosin, original magnification x40). b) The cells express S-100 protein, original magnification x100. |
Discussion
Eyelid schwannoma is a rare, slowly growing, benign neoplasm. Schwann cell tumors generally arise from peripheral or cranial nerves. Macroscopically, they appear to be well demarcated but not encapsulated; they usually grow very slowly and are asymptomatic. There are several clinico-pathologic variants of schwannoma, including conventional schwannoma, cellular schwannoma, and melanotic schwannoma [7]. Microscopically, they may demonstrate a biphasic pattern with areas of highly cellular (Antoni type A) and myxoid matrix (Antoni type B) [7]. The most important feature in its diagnosis remains the strong reactivity to S100 protein by immunohistochemistry, particularly in Antoni type A areas. Despite sometimes striking cytologic atypia, mitotic figures are rare. It is postulated that degenerative changes occur due to the long period of time over which large schwannomas develop [8].
Because of their rarity and location, eyelid schwannomas have been confused with epidermal inclusion cysts. Their management is complete excision with clear margins to establish the histopathologic diagnosis and prevent recurrence. Incomplete removal is associated with eventual recurrence and more aggressive behavior [4,6]. An attempt to preserve the continuity of the nerve should be made, but this is not always possible and does not appear to have any major consequences in this site. Histopathologically, the neoplasm appears to be well-circumscribed and composed of bundles of elongated cells with thin, attenuated nuclei. These cells tend to align themselves into compact parallel rows, with intermittent dense anucleate zones. In other locations, a poor prognosis has been described if the cells are fusiform, contain melanin granules, or if epithelioid cells are present [2]. Nevertheless, malignant transformation has not been reported in eyelid schwannomas.
In our Department of Ophthalmology, schwannomas represent 0.1 percent (2/2400) of the eyelid neoplasms in a period of eleven years (1995-2005). Both cases were described as conventional schwannoma. The age range in the adult group of published cases (in 40 years) was between 19 and 63. The female to male proportion was 3:2. Our cases were not associated with neurofibromatosis. In case 1, the mass was located on the middle section of the eyelid and involved the edge of the upper eyelid. This location has not been described previously in adults, and the tumor probably arose from branches of the supraorbital nerve. In case 2, the mass was located on the middle section of the lower eyelid and was subcutaneous, probably arising from branches of the infraorbital nerve.
Schwannomas of ophthalmic interest are rare, although they have been reported in relation to the orbit (1% of orbital tumors), and infrequently to the uveal tract and conjunctiva. Eyelid schwannomas are extremely uncommon. Although very rare, schwannoma should be taken into consideration in the differential diagnosis of any eyelid neoplasm.
References
1. Baijal GC, Garg SK, Kanhere S, Monga S. Schwannoma of the eye-lid. Indian J Ophthalmol. 1980; 28:155-156.2. Butt Z, Ironside JW. Superficial epithelioid schwannoma presenting as a subcutaneous upper eyelid mass. Br J Ophthalmol. 1994; 78:586-588.
3. Reeh MJ. Treatment of Lid and Epibulbar Tumors. Springfield, IL: Thomas Publishers; 1963.
4. Shields JA, Guibor P. Neurilemoma of the eyelid resembling a recurrent chalazion. Arch Ophthalmol. 1984; 102:1650.
5. Siddiqui MA, Leslie T, Scott C, MacKenzie J. Eyelid schwannoma in a male adult. Clin Experiment Ophthalmol. 2005; 33:412-413.
6. Shields JA, Kiratli H, Shields CL, Eagle RC Jr, Luo S. Schwannoma of the eyelid in a child. J Pediatr Ophthalmol Strabismus. 1994; 31:332-333.
7. Scheithauer BW, Woodruff JM, Erlandson RA. Tumors of the peripheral nervous system. In: Rosai J, Sobin LH (eds) Atlas of Tumor Pathology. Third series, fasc. 24, Armed Forces Institute Pathology: Washington, D.C., pp105-176; 1999.
8. Dahl I. Ancient neurilemmoma (Schwannoma). Acta Pathol Microbiol Scand. 1977; 85:812-818.
© 2007 Dermatology Online Journal